
Abstract
Purpose
The molecular mechanism of breast and/or ovarian cancer susceptibility remains unclear in the majority of patients. While germline mutations in the regulatory non-coding regions of BRCA1 and BRCA2 genes have been described, screening has generally been limited to coding regions. The aim of this study was to evaluate the contribution of BRCA1/2 non-coding variants.
Methods
Four BRCA1/2 non-coding regions were screened using high-resolution melting analysis/Sanger sequencing or next-generation sequencing on DNA extracted from index cases with breast and ovarian cancer predisposition (3926 for BRCA1 and 3910 for BRCA2). The impact of a set of variants on BRCA1/2 gene regulation was evaluated by site-directed mutagenesis, transfection, followed by Luciferase gene reporter assay.
Results
We identified a total of 117 variants and tested twelve BRCA1 and 8 BRCA2 variants mapping to promoter and intronic regions. We highlighted two neighboring BRCA1 promoter variants (c.-130del; c.-125C > T) and one BRCA2 promoter variants (c.-296C > T) inhibiting significantly the promoter activity. In the functional assays, a regulating region within the intron 12 was found with the same enhancing impact as within the intron 2. Furthermore, the variants c.81-3980A > G and c.4186-2022C > T suppress the positive effect of the introns 2 and 12, respectively, on the BRCA1 promoter activity. We also found some variants inducing the promoter activities.
Conclusion
In this study, we highlighted some variants among many, modulating negatively the promoter activity of BRCA1 or 2 and thus having a potential impact on the risk of developing cancer. This selection makes it possible to conduct future validation studies on a limited number of variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At least 10% of the 14 million breast cancer diagnoses made worldwide each year are associated with hereditary predisposition. Breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2) are the two most penetrant genes implicated in hereditary breast and/or ovarian cancer (HBOC) [1, 2]. However, a causal mutation useful for genetic counseling is identified in less than 15% of tested families and, in most cases, little is known about the underlying molecular mechanisms of cancer susceptibility. It would be particularly useful to identify inherited mutations in patients with a family history of cancers to allow implementation of risk reduction strategies for these patients and their families. New technologies have been proposed to study a panel of genes known or suspected to be involved in breast and/or ovarian cancer predisposition. Other HBOC predisposition genes have also been explored but could represent less than 5% of all causative mutations [3]. BRCA1/2 coding variants remain the major contributors to HBOC risk and the hypothesis that the remaining predisposition is also related to these genes remains plausible and could be explained by the presence of variants in non-coding regions for which the functional impact is currently unknown.
Progressing sequencing technologies and the development of bioinformatics tools now allow more informed exploration of transcriptional regulation [4, 5]. Germline mutations in the regulatory regions of the genome may represent an important tumorigenic mechanism and the impact of some non-coding regions on transcription regulation of the BRCA1/2 genes has already been reported. Large genomic deletions involving the BRCA1 and BRCA2 promoters increase the risk of cancer [6,7,8]. Wardrop et al. described two non-coding sequences in intron 2 located 2.5 kb downstream to the BRCA1 promoter with differential transcriptional regulatory activity [9]. Germline variants in the BRCA1 and BRCA2 5′ and 3′UTRs, resulting in reduced translation efficiency, have also been described [10,11,12,13,14,15]. Moreover, several examples of variations in the non-coding sequences of other genes have also been correlated with cancer risk. Recently, two different recurrent mutations in the promoter of the telomerase reverse transcriptase (TERT) gene generating telomerase overexpression have been demonstrated to be associated with an increased risk of melanoma [16].
A reasonable mechanism to explain the impact of alterations of non-coding sequences on cancer risk is that the nucleotide change can create or disrupt a binding motif for a given transcription factor, and consequently alter the protein expression in all tissues expressing this factor. However, there is currently a lack of information about the function and polymorphisms of non-coding sequences and genetic screening of BRCA1/2 genes is generally limited to coding regions and intron–exon junctions. The role of variants in non-coding regions with no splicing effect has not been thoroughly investigated and even less is known about their contribution to transcriptional regulation. Assessment of their impact on cancer predisposition is often more complex. The present study is a first approach to provide data to allow estimations of the impact of these variants on breast and/or ovarian cancer risk.
The primary objective of this study was to assess the significance and contribution of non-coding variants on BRCA1/2 promoter activity and on breast and/or ovarian cancer risk.
Materials and methods
DNA samples, probands, and cohorts
In order to identify novel germline mutations that could explain hereditary predisposition, patients from three different HBOC cohorts, with eligibility criteria for familial genetic testing according to the French consensus statement and negative for BRCA1/2 causal mutation, were enrolled [17–19]. A total of 1968 patients were tested at Centre François Baclesse, Caen, 1958 patients were tested at Institut Curie, Saint-Cloud, and 723 patients were tested at Institut Curie, Paris (Fig. 1, Table 1A). The characteristics of each cohort have been previously described [3, 20,21,22]. The frequency of the variants identified was also evaluated in a control cohort composed of Institut Curie patients with a cancer predisposition other than breast or ovarian cancer. The analysis was done anonymously and the frequency of the variant was only reported to compare with the cases.

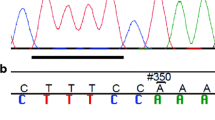
a Work flow diagram describing the screening strategy and variant prioritization. b Location of the non-coding regions studied and the respective variants of each region selected for functional analysis
DNA was extracted from lymphoblastoid cell lines and 4 BRCA1/2 non-coding regions were screened by HRM or NGS: BRCA1 promoter, BRCA1 intron 2, BRCA1 intron 12, and BRCA2 promoter (Fig. 1, Table 1B).
In addition to the variants identified by this screening, we also selected new variants from the ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) database [23], in the context of a collaborative study.
Screening of BRCA1/2 non-coding regions: high-resolution melting analysis and next-generation sequencing
The four regions explored had been previously defined as being regions most likely to be functional and presenting a higher probability of containing disease-associated variants. This analysis comprised bioinformatics, experimental and population-based approaches to identify and validate key non-coding regions in BRCA1 and BRCA2 [9, 24, 25]. For example, the regions explored in introns 2 and 12 are highly conserved among mammalian species and contain many potential binding sites for known transcription factors [9].
For HRM screening, PCR reactions were performed in duplicate in a final volume of 15 μl containing 2 ng of DNA, 0.6 μM of each primer (forward or reverse), 1 × LightCycler 480 HRM Master mix (Roche), and LightCycler® 480 Resolight Dye or LCGeen® Plus melting dye for BRCA1 and BRCA2 screening, respectively [26]. Each assay included DNA with known BRCA1/2 mutation corresponding to the primer set as positive control. The PCR program is available on demand. The non-coding BRCA1/2 DNA sequences evaluated and the primers selected for this purpose are specified in Supplementary Table 1.
NGS screening was performed with a dedicated panel for cancer predisposition with Illumina sequencers [3, 21, 22]. All known genetic variants detected were confirmed by sequencing PCR products (Sanger sequencing method).
In silico analysis and variant prioritization
For variant prioritization, we first applied a population frequency filter to exclude variants with an allele frequency > 1%. The minor allelic frequency (MAF) was estimated from the Ensembl project or Exome Aggregation Consortium [27, 28]. Information analysis was then performed to identify potentially pathogenic variants. This approach evaluates the effects of the variant on binding sites and whether the variant involves the creation, strengthening, weakening, or abolition of a binding site [5].
All variants were scanned with Shannon Human Splicing Mutation Pipeline, a genome-scale analysis program that predicts the effects of variants on mRNA splicing [29]. Variants were selected according to the following criteria: weakened natural site ≥ 1.0 bits or strengthened cryptic site equal to or greater than the nearest natural site of the same phase. We also analyzed the effects of variants in the 5′UTR region on TF binding using the models previously described by Mucaki et al. [5].
Finally, for functional assays, we prioritized variants located in domains most likely to be functional based on bioinformatics analysis, and for which testing tools were available.
Luciferase reporter gene constructions
Luciferase reporter plasmids containing sequences from the BRCA1 promoter and BRCA1 intron 2 have been described previously [9, 25]. For the BRCA2 luciferase reporter plasmid, a 750 bp region containing the BRCA2 promoter was cloned into the pGL3-Basic vector [9, 25]. In these plasmids, promoter sequences were inserted upstream to the coding sequence of firefly luciferase in the XhoI site. The intronic sequences were inserted immediately downstream to the luciferase gene in the BamH1 site (Fig. 2). A new construct was made in order to clone a region of BRCA1 intron 12 downstream to the luciferase gene, using the Gibson Assembly Method [30]. Variants were introduced into the plasmids by directed mutagenesis. BRCA1: c.-287C > T and c.-326_324del variants were used as positive controls. As the BRCA2 promoter has been less studied, it was not possible to model a positive control for it, and thus the wild-type promoter was used as a reference. The BRCA2: c.-52A > G polymorphism was used as negative control. All constructs were verified by DNA sequencing.

Representation of the plasmids used in this study. a BRCA2 promoter. b BRCA1 promoter. c BRCA1 promoter and BRCA1 intron 2. d BRCA1 promoter and BRCA1 intron 12
Cell culture, transfection, and dual-luciferase reporter assay
The triple-negative breast cancer MDA-MB-231 cell line and the estrogen receptor-positive MCF-7 breast cancer cell line were obtained from American type culture collection (ATCC). MDA-MB-231 was used in every experiment. We confirmed some of the significant results in the MCF-7 breast cancer cell line. All cells were tested regularly for mycoplasma contamination using plasma Test (invivoGen) and authenticated using the GenePrint 10 system Kit (Promega). MCF-7 and MDA-MB-231 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and antibiotics (37 °C, 5% CO2). To perform transient transfection, cells were seeded in 24-well plates and were subsequently transfected at 80% confluence using X-treme (QIAGEN) reagent according to the manufacturer’s instructions. After 36 h, Firefly and Renilla activities were measured using the dual-luciferase kit (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity and expressed as mean ± S.D. of triplicates from a representative experiment.
All statistical calculations were performed using PASW Statistics (version 18.0; SPSS Inc., Chicago, IL). Comparisons were performed using a two-sided unpaired Student t test. p values less than 0.05 were considered to be statistically significant.
Clinico-pathological features of variant carriers
When a significant reduction of promoter activity was observed, more evidence for variant classification was sought. Further analysis of the patient’s pedigree, allelic imbalance in RNA transcription, and tumor sample features, including Loss of Heterozygosity (LOH) and methylation, were determined, when material was available. LOH analysis was performed by Sanger sequencing or pyrosequencing. The BRCA1 promoter methylation status was also assessed for variants with functional impact and when the material was available by pyrosequencing assay [31].
Results
Identification of new variants in BRCA1/2 non-coding regions
The aim of this study was to identify novel germline mutations located in the non-coding regions of BRCA1 and BRCA2 genes that could explain hereditary predisposition for breast cancer. To do this, 4 BRCA1/2 regions of the DNA of patients from 3 different HBOC cohorts were screened: BRCA1 promoter, BRCA1 intron 2, BRCA1 intron 12, and BRCA2 promoter (Table 1B). This approach allowed the identification of 117 variants in BRCA1/2 non-coding regions (Fig. 1, Tables 1A, Supplementary Tables 2 and 3).
Five of these 117 variants were identified in more than 4 families: c.81-3625del, c.-20 + 11C > T, c.4186-2050A > G and c.-86C > T in BRCA1 gene, and c.-175C > T in BRCA2 gene. Two of them were found exclusively in our cohorts with HBOC predisposition: c.81-3625del and c.-20 + 11C > T in BRCA1 gene. The remaining three variants were also identified in the control population.
In silico analyses
In silico analysis of these 117 variants identified 3 BRCA1 variants with a potential impact on splicing: c.-73C > G, c.-86C > T, and c.-19-130insA; 3 BRCA1 variants with a potential impact on UTR binding site alteration: c.-73C > G, c.-79G > T, and c.-121G > C; and twelve BRCA1 variants with a potential impact on the TFB site: c.81-3459C > T, c.81-3510C > T, c.-19-479G > T, c.-20 + 131delGGCGTA, c.-20 + 131A > T, c.-20 + 125A > C, c.-177C > T, c.-130del, c.-125C > T, c.-20 + 486insG, c.-19-123insAT, and c.-20 + 11C > T. The impact of the variants on RNA secondary structure was also analyzed and one BRCA1 variant, c.-130del, displayed a predicted impact on mRNA conformation (Fig. 3).

BRCA1 variant: c.-130del—structure with mFOLD is significantly changed due to loss of C-G bond
Moreover, two variants in intron 2 of BRCA1 could have an impact on the creation of cryptic exons: c.81-4118G > A and c.81-3519G > T. Validation of these cryptic exons would require the development of a dedicated RT-PCR on mRNA. No suspected mRNA splicing effect was detected in silico for these variants.
Six BRCA2 variants were identified with different potential impacts: c.-112G > A (UTR binding site and splicing factor binding site), c.-123G > A (splicing factor binding site), c.-171G > C (mRNA structure), c.-178insCTGCTGCGCCT (TFB site), c.-213G > T (UTR binding site), c.-296C > T (TFB site). The c.-171G > C variant also displayed a predicted impact on mRNA structure.
Based on these analyses and taking into account the available tools, twenty variants were selected for functional assays [32]. Nine of these 20 variants were located in the BRCA1 promoter region, two variants were located in BRCA1 intron 2, one variant was located in BRCA1 intron 12, and eight variants were located in the BRCA2 promoter region (Table 2).
Impact of variants on BRCA2 promoter activity
Among the 8 BRCA2 variants tested, only c.-296C > T induced a significant reduction (28%) of reporter gene expression, indicating that this variant inhibits the BRCA2 promoter activity (Fig. 4). Moreover, analysis of the tumor sample harboring this variant identified LOH of the wild-type allele, and the patient’s pedigree revealed that one of her 2 sisters had also a diagnosis of breast cancer at the age of 44 years (Table 3, F1), further supporting the potential pathogenic impact of this variant (Fig. 1 supplementary data).

Impact of different variants on BRCA2 promoter activity. MDA-MB-231 breast cell line was transfected with the expression vector pRL-TK Renilla in combination with the luciferase reporter plasmids containing the BRCA2 promoter wild type (Promoter WT) or possessing a variant as indicated. Twenty-four hours later, cell extracts were prepared and luciferase activities quantified
Two variants showed an increase of promoter activity: the eventual role of this positive effect on cancer remains to be defined. The other variants demonstrated similar levels of activity to that of the wild-type sequence strongly suggesting that these variants are neutral (Fig. 4).
Impact of variants on BRCA1 promoter activity
The BRCA1 variants analysis revealed two neighboring variants: c.-125C > T and c.-130del, inducing a strong reduction of promoter activity (60% reduction for c.-130del p = 0.0002, and 56% reduction for c.-125C > T p = 0.0025) (Fig. 5 and Table 2B). To confirm these results, we repeated the experiment in another breast cancer cell line, MCF-7. We validated our first results (70% reduction for c.-130del, p = 0.003, and 30% reduction for c.-125C > T, p = 0.003) (Table 2B). One family was available for the BRCA1 c.-130del with many prostate cancers (Table 3, F2). As for the BRCA2 promoter, we also found 2 variants increasing weakly the BRCA1 promoter activity: c.-362T > G; c.-121 G > C (Fig. 5 and Table 2A). The remaining variants were associated with similar reporter gene activity to that of the wild-type sequence (Fig. 5).

Impact of different variants on BRCA1 promoter activity. MDA-MB-231 breast cell line was transfected with the expression vector pRL-TK Renilla in combination with the luciferase reporter plasmids containing the BRCA1 promoter wild type (Promoter WT) or possessing a variant as indicated. Twenty-four hours later, cell extracts were prepared and luciferase activities quantified. The c.-287C > T and c.-326_324del variants are artificial constructions on CAAT box and on the RIBS element, respectively, used as positive controls
We also studied the impact of BRCA1 intronic variants on BRCA1 promoter activity: two detected in intron 2 (c.81-3985A > T and c.81-3980A > G) and one detected in intron 12(c.4186-2022C > T). First of all, we confirmed that the presence of a part of intron 2 and also a part of intron 12 increased the activity of the BRCA1 promoter, 1.48- and 1.72-fold, respectively, confirming that these two introns possess important regulatory sequences (Fig. 6a). The intron 2 effect was already described contrary to the intron 12 [9]. The intronic variant c.81-3985A > T is located in a repressor region previously described in intron 2 [9]. However, we did not detect any influence of this variant on the positive effect of the intron 2 on the BRCA1 promoter activity. Most importantly, we found that in the presence of the two intronic variants (c.81-3980A > G and c.4186-2022C > T), the introns 2 and 12 had no longer an impact over BRCA1 promoter activity (Fig. 6b and Table 2B).

a Impact of different intronic variants on BRCA1 promoter activity. MDA-MB-231 breast cell line was transfected with the expression vector pRL-TK Renilla in combination with the luciferase reporter plasmids containing the BRCA1 promoter wild type without (Promoter WT) or with the intron 2 or 12 wild type (a) or possessing a variant (b) as indicated. Twenty-four hours later, cell extracts were prepared and luciferase activities quantified
We did not detect any BRCA1 promoter methylation for any functionally active variants.
Discussion
Results statement
Optimal management of hereditary breast and/or ovarian cancer families requires accurate identification of individuals at genuinely high risk. Although it is important to identify new breast and ovarian cancer susceptibility genes, non-coding regions are currently not investigated, with the exception of those intronic variants with an impact on RNA splicing [33, 34]. In the present study, we chose to explore these non-coding regions and carry out functional assays for these variants. Screening of the HBOC population comprising 3926 patients screened for BRCA1 and 3010 patients screened for BRCA2 non-coding regions revealed 117 variants (0.5 to 1.4% of the screened population).
We have validated an experimental protocol for the initial functional classification of 20 of these variants that demonstrated 10 non-coding variants with a functional impact on BRCA1/2 promoter activity. Among these 10 variants, two decreased BRCA1 promoter activity: c.-130del and c.-125C > T; one decreased BRCA2 promoter activity: c.-296C > T; and two (c.81-3980A > G and c.4186-2022C > T) suppressed the positive effect of the introns 2 and 12 over the BRCA1 promoter activity.
Limitations of functional assays for non-coding variants
Fluctuations of the basal reporter activity were observed for both the BRCA1 and BRCA2 promoters, which could be explained by poorly controlled parameters of the biological system as well as technical limitations, for example, the quality and conformation of transfected DNA. An internal positive control was always used to ensure correct interpretation of functional results. It is noteworthy that only minor differences were observed for PGL3 basic or Renilla luciferase activity, which confirm transfection efficiency, and that the wild-type promoter was always present to ensure correct interpretation of functional results. Moreover, the results for the potential suppressor variants, BRCA1 c.-125C > T; BRCA1 c.-130del; BRCA2 c.-296C > T, were always consistent under the various experimental conditions.
Sensitive region in promoter of BRCA1
We identified a sensitive region in the BRCA1 promoter with 3 functionally active variants: c.-125C > T; c.-130del; c.-121G > C, including 2 with a marked repressor impact on promoter activity (Fig. 7). Analysis of the DNA sequence region containing the neighboring BRCA1 c.-125C > T and BRCA1 c.-130del promoter variants, using the Swiss Regulon TF database (http://swissregulon.unibas.ch/), revealed that both variants are located in a putative E2F1 transcription factor binding site (TFBS)(Fig. 7). These two variants may thus impact the ability of E2F1 to induce BRCA1 transcription. An E2F1 information model generated using ChIP-Seq data from HeLa-S3 lysates revealed a fairly weak 3.6 bit E2F1 site on the negative strand (Fig. 7a) [35]. When the binding site was analyzed from the negative strand (the orientation of BRCA1 transcription), both mutations were predicted to decrease the strength of the predicted E2F1 site. Variant c.-125C > T was predicted to be a weak variant mainly due to the presence of a ‘T’ in its sequence when a C or G was expected (TGCGCG; arrow indicates the position of T relative to our model; Fig. 7a). Our analysis also revealed that the c.-130del variant is located in a putative HSF1 and TEAD4 TFBSs. Other transcription factors identified in future studies could therefore increase our understanding of the biological implications of these variants in TFBSs.

Identification of a new potential E2F1 binding site in BRCA1 promoter. a Information Models built from publically available ChIP-Seq data (HeLa-S3). b Models from SwissRegulon (Fig. 5a)
Our in silico analysis revealed that the BRCA1: c.-130del variant also has a potential impact on the RNA 2D structure. The RNA conformation of the first exon of the BRCA1 gene has been described and could have an impact on transcription, as the alternative exon 1b transcript of the BRCA1 gene has a conformation that could reduce translation of mRNA [36]. This impact cannot be detected with the luciferase assay.
Analysis of the pedigree of the c.-130del index case, looking for more evidence for classification of variants, revealed numerous cases of prostate cancer, usually associated with alterations of the BRCA2 gene. Patients carrying a BRCA1 mutation usually present little or no increased cancer risk, but a more aggressive form of prostate cancer [37]. Unfortunately, sequencing of this patient’s tumor sample did not reveal any additional useful for classification: neither LOH of the wild-type allele nor promoter methylation was detected. However, recent studies have demonstrated the effect of BRCA1-haploinsufficiency in various cells and tissues, which may explain how mutation in a single BRCA1 allele conferred increased cancer risk in this patient [38].
BRCA2 promoter
For the first time, a variant of the BRCA2 promoter has been shown to have a functional impact on transcription (c.-296C > T). This variant is also located close to a region rich in transcription factor binding sites. Analysis of the tumor sample from a carrier of this variant revealed somatic loss of the wild-type BRCA2 allele, suggesting that loss of heterozygosity may play a role in the tumorigenesis. The other two BRCA2 variants (c.-280_-272dup and c.-123G > A) showed an enhancer activity, the consequence of which is unknown.
Putative changes in TFBS related to the presence of the variants
The two BRCA2 variants with a significant impact on transcription (c.-296C > T and BRCA2: c.-280_-272dup) were correlated with the TFBS predictions based on the variant prioritization method (Table 2A). These variants alter the binding strength of two PAX5 binding sites. ChIP-Seq experiments have shown that PAX5 binds to the BRCA2 promoter region. Furthermore, although the PAX5 gene has not been shown in the literature to have a direct effect on BRCA expression, it has been shown to be hypermethylated in triple-negative breast cancer [39]. Loss of a PAX5 binding site may therefore induce a similar effect to that of an overall reduction of PAX5 gene expression.
TFBS analysis showed weakening of PAX5 binding site from 12.7 to 8.5 bits in the presence of the c.-296C > T variant. Similarly, the promoter activity assay showed an increase in BRCA2 promoter activity in the presence of the BRCA2: c.-280_-272dup event. TFBS analysis predicted that this duplication would create a 5.6 bit PAX5 binding site, which correlates with the reported increase in promoter activity.
Introns 2 and 12 BRCA1
Wardrop et al. have described the presence of regulatory regions in the intron 2 sequence of BRCA1 gene [9]. Although these regions are situated several kb downstream to the promoter region, they regulate BRCA1 expression at the transcriptional level, most likely via gene looping [25]. We investigated introns 2 and intron 12. Intron 12 locus has been selected for being rich on the transcription factor binding sites and interspecies conservation.
Even if the variant c.81-3985A > T was found in three families (Table 3) suspected for cancer predisposition, we did not detect any influence of this variant on the positive effect of the intron 2 over the BRCA1 promoter activity. This result strongly suggest that the c.81-3985A > T variant do not inhibit the activity of the BRCA1 promoter and therefore would have no effect on the breast cancer development. Furthermore, analysis of RNA from the patient’s lymphoblastoid cell line showed no allelic imbalance, which support our conclusion that the c.81-3985A > T variant may have no causal impact on cancer (data not shown).
In the other hand, we found that the two intronic variants c.81-3980A > G and c.4186-2022C > T displayed wild-type devoid of intron 2 or 12, respectively. These two variants may inhibit BRCA1 promoter activity by suppressing the positive effect of the intron 2/12 on the BRCA1 promoter activity thereby stimulating cancer development. In this study, the regulating impact of intron 12 has been confirmed in vitro and this work highlights the importance of screening this region. Some variants were identified and a variant c.4186-2022C > T has been able to revert the enhancing impact of the intron 12 locus. Unfortunately, there was no material available to work on these variants.
Epigenetics
It is difficult to draw any solid conclusions from these results that could be used for genetic counseling of carriers of variants in BRCA1/2 non-coding regions. Constitutional epimutation of the promoter has been described for the MLH1 gene with a cis-acting variant, and a relationship between promoter activity and level of methylation has been established [40,41,42]. All of these cases presented somatic mosaicism between tissues and family members. No epimutations have been reported in the BRCA2 gene. However, the promoter of BRCA1 gene can also be methylated and constitutional epimutations have been reported [43]. No methylation of the promoter was identified on the c.-130del variant.
Conclusion
This study put in evidence the presence of rare variants in the non-coding regions of the BRCA1 and BRCA2 genes, and 5 of them induced a significant reduction of transcriptional levels. Our data raise the question whether the presence of these variants in regulatory regions may have an impact on the risk of developing cancer. To be more conclusive, it would be helpful to obtain more information about the frequency of these alterations. The model including the functional assay here described can be a useful tool to highlight the variants requiring further investigation including epimutation or co-segregation analysis, in order to ultimately establish a potential association with cancer risk.
References
Miki Y, Swensen J, Shattuck-Eidens D et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71
Wooster R, Bignell G, Lancaster J et al (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378:789–792. https://doi.org/10.1038/378789a0
Castéra L, Krieger S, Rousselin A et al (2014) Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet EJHG 22:1305–1313. https://doi.org/10.1038/ejhg.2014.16
Caminsky NG, Mucaki EJ, Perri AM et al (2016) Prioritizing variants in complete Hereditary Breast and Ovarian Cancer (HBOC) genes in patients lacking known BRCA mutations. Hum Mutat 37:640–652. https://doi.org/10.1002/humu.22972
Mucaki EJ, Caminsky NG, Perri AM et al (2016) A unified analytic framework for prioritization of non-coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Med Genom 9:19. https://doi.org/10.1186/s12920-016-0178-5
Puget N, Stoppa-Lyonnet D, Sinilnikova OM et al (1999) Screening for germ-line rearrangements and regulatory mutations in BRCA1 led to the identification of four new deletions. Cancer Res 59:455–461
Brown MA, Lo L-J, Catteau A et al (2002) Germline BRCA1 promoter deletions in UK and Australian familial breast cancer patients: identification of a novel deletion consistent with BRCA1:psiBRCA1 recombination. Hum Mutat 19:435–442. https://doi.org/10.1002/humu.10055
Walsh T, Casadei S, Coats KH et al (2006) Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295:1379–1388. https://doi.org/10.1001/jama.295.12.1379
Wardrop SL, Brown MA, kConFab Investigators (2005) Identification of two evolutionarily conserved and functional regulatory elements in intron 2 of the human BRCA1 gene. Genomics 86:316–328. https://doi.org/10.1016/j.ygeno.2005.05.006
Wang J, Lu C, Min D et al (2007) A mutation in the 5′ untranslated region of the BRCA1 gene in sporadic breast cancer causes downregulation of translation efficiency. J Int Med Res 35:564–573
Marino M, Rabacchi C, Simone ML et al (2009) A novel deletion of BRCA1 gene that eliminates the ATG initiation codon without affecting the promoter region. Clin Chim Acta 403:249–253. https://doi.org/10.1016/j.cca.2009.02.020
Pongsavee M, Yamkamon V, Dakeng S et al (2009) The BRCA1 3′-UTR: 5711 + 421T/T_5711 + 1286T/T genotype is a possible breast and ovarian cancer risk factor. Genet Test Mol Biomark 13:307–317. https://doi.org/10.1089/gtmb.2008.0127
Lheureux S, Lambert B, Krieger S et al (2011) Two novel variants in the 3′UTR of the BRCA1 gene in familial breast and/or ovarian cancer. Breast Cancer Res Treat 125:885–891. https://doi.org/10.1007/s10549-010-1165-8
Pelletier C, Speed WC, Paranjape T et al (2011) Rare BRCA1 haplotypes including 3′UTR SNPs associated with breast cancer risk. Cell Cycle Georget Tex 10:90–99. https://doi.org/10.4161/cc.10.1.14359
Pamuła J, Krześniak M, Zientek H et al (2006) Functional impact of sequence alterations found in BRCA1 promoter/5′UTR region in breast/Ovarian Cancer Families from Upper Silesia, Poland. Hered Cancer Clin Pract 4:20–24. https://doi.org/10.1186/1897-4287-4-1-20
Horn S, Figl A, Rachakonda PS et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339:959–961. https://doi.org/10.1126/science.1230062
Eisinger F, Alby N, Bremond A et al (1999) Inserm ad hoc committee: recommendations for the management of women with a genetic risk for developing cancer of the breast and/or the ovary. Bull Cancer 86:307–313 (Paris)
Eisinger F, Bressac B, Castaigne D et al (2006) Identification and management of hereditary breast-ovarian cancers (2004 update). Pathol Biol 54:230–250. https://doi.org/10.1016/j.patbio.2006.02.002 (Paris)
Eisinger F, Bressac B, Castaigne D et al (2004) Identification and management of hereditary predisposition to cancer of the breast and the ovary (update 2004). Bull Cancer 91:219–237 (Paris)
Caputo S, Benboudjema L, Sinilnikova O et al (2012) Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res 40:D992–D1002. https://doi.org/10.1093/nar/gkr1160
Tarabeux J, Zeitouni B, Moncoutier V et al (2014) Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur J Hum Genet EJHG 22:535–541. https://doi.org/10.1038/ejhg.2013.181
Collet A, Tarabeux J, Girard E et al (2015) Pros and cons of HaloPlex enrichment in cancer predisposition genetic diagnosis. Genet 2:263–280. https://doi.org/10.3934/genet.2015.4.263
Spurdle AB, Healey S, Devereau A et al (2012) ENIGMA—evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat 33:2–7. https://doi.org/10.1002/humu.21628
Saunus JM, French JD, Edwards SL et al (2008) Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res 68:9469–9478. https://doi.org/10.1158/0008-5472.CAN-08-1159
Tan-Wong SM, French JD, Proudfoot NJ, Brown MA (2008) Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc Natl Acad Sci USA 105:5160–5165. https://doi.org/10.1073/pnas.0801048105
Coulet F, Pires F, Rouleau E et al (2010) A one-step prescreening for point mutations and large rearrangement in BRCA1 and BRCA2 genes using quantitative polymerase chain reaction and high-resolution melting curve analysis. Genet Test Mol Biomark 14:677–690. https://doi.org/10.1089/gtmb.2009.0183
Consortium EA, Lek M, Karczewski K, et al (2015) Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 030338. https://doi.org/10.1101/030338
Aken BL, Ayling S, Barrell D et al (2016) The Ensembl gene annotation system. Database. https://doi.org/10.1093/database/baw093
Shirley BC, Mucaki EJ, Whitehead T et al (2013) Interpretation, stratification and evidence for sequence variants affecting mRNA splicing in complete human genome sequences. Genom Proteom Bioinform 11:77–85. https://doi.org/10.1016/j.gpb.2013.01.008
Gibson DG, Young L, Chuang R-Y et al (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. https://doi.org/10.1038/nmeth.1318
Tost J, Gut IG (2007) DNA methylation analysis by pyrosequencing. Nat Protoc 2:2265–2275. https://doi.org/10.1038/nprot.2007.314
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20:110–121. https://doi.org/10.1101/gr.097857.109
Anczuków O, Buisson M, Léoné M et al (2012) BRCA2 deep intronic mutation causing activation of a cryptic exon: opening toward a new preventive therapeutic strategy. Clin Cancer Res Off J Am Assoc Cancer Res 18:4903–4909. https://doi.org/10.1158/1078-0432.CCR-12-1100
Garcia AI, Buisson M, Damiola F et al (2016) Mutation screening of MIR146A/B and BRCA1/2 3′-UTRs in the GENESIS study. Eur J Hum Genet EJHG. https://doi.org/10.1038/ejhg.2015.284
Lu R, Mucaki EJ, Rogan PK (2017) Discovery and validation of information theory-based transcription factor and cofactor binding site motifs. Nucleic Acids Res 45:e27. https://doi.org/10.1093/nar/gkw1036
Xu CF, Brown MA, Chambers JA et al (1995) Distinct transcription start sites generate two forms of BRCA1 mRNA. Hum Mol Genet 4:2259–2264
Gallagher DJ, Gaudet MM, Pal P et al (2010) Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res 16:2115–2121. https://doi.org/10.1158/1078-0432.CCR-09-2871
Staff S, Isola J, Tanner M (2003) Haplo-insufficiency of BRCA1 in sporadic breast cancer. Cancer Res 63:4978–4983
Hafez MM, Al-Shabanah OA, Al-Rejaie SS et al (2015) Increased hypermethylation of glutathione S-transferase P1, DNA-binding protein inhibitor, death associated protein kinase and paired box protein-5 genes in triple-negative breast cancer Saudi females. Asian Pac J Cancer Prev APJCP 16:541–549
Ward RL, Dobbins T, Lindor NM et al (2013) Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med Off J Am Coll Med Genet 15:25–35. https://doi.org/10.1038/gim.2012.91
Gylling A, Ridanpää M, Vierimaa O et al (2009) Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer 124:2333–2340. https://doi.org/10.1002/ijc.24230
Hesson LB, Packham D, Kwok C-T et al (2015) Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression. Hum Mutat 36:622–630. https://doi.org/10.1002/humu.22785
Hansmann T, Pliushch G, Leubner M et al (2012) Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet 21:4669–4679. https://doi.org/10.1093/hmg/dds308
Acknowledgements
The authors thank the French oncogeneticists, the UNICANCER Genetic Group leads by Dr Catherine Nogues, and probands for their cooperation. This work was supported by the Association pour la Recherche en Cancérologie de Saint-Cloud (ARCS), by the National Cancer Institute (INCa: INCA-DGOS_8706) and by the National Health and Medical Research Council (Australia) Grant #1104808. We gratefully acknowledge Dr Lisa Golmard for her help during the patients screening.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
dos Santos, E.S., Caputo, S.M., Castera, L. et al. Assessment of the functional impact of germline BRCA1/2 variants located in non-coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Res Treat 168, 311–325 (2018). https://doi.org/10.1007/s10549-017-4602-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4602-0