
Abstract
Plant height is an important plant architecture trait that determines the canopy structure, photosynthetic capacity and lodging resistance of upland cotton populations. To understand the genetic basis of plant height for marker-assisted breeding, quantitative trait loci (QTL) analysis was conducted based on the genetic map of recombinant inbred lines (RILs) derived from the cross “CRI12 × J8891” (Gossypium hirsutum L.). Three methods, including composite interval mapping, multiple interval mapping and multi-marker joint analysis, were used to detect QTL across multiple environments in the RILs and in the immortalized F2 population developed through intermating between RILs. A total of 19 QTL with genetic main effects and/or genetic × environment interaction effects were identified on 15 chromosomes or linkage groups, each explaining 5.8–14.3 % of the phenotypic variation. Five digenic epistatic QTL pairs, mainly involving additive × additive and/or dominance × dominance, were detected in different environments. Seven out of eight interacting loci were main-effect QTL, suggesting that these loci act as major genes as well as modifying genes in the expression of plant height. The results demonstrate that additive effects, dominance and epistasis are all important for the genetic constitution of plant height, with additive effects playing a more important role in reducing plant height. QTL showing stability across environments that were repeatedly detected by different methods can be used in marker-assisted breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant height, which is associated with plant morphogenesis and lodging resistance, is an important agronomic trait that has been used in plant architecture breeding to achieve yield potential in crops. The introduction of dwarfing genes into wheat and rice successfully increased the harvest index, and thus grain yield, during the “Green Revolution” (Evans 1993; Gale et al. 1985). Unraveling the genetic basis of plant height has long been a target of plant research. In recent years, with the development of molecular markers and mapping technology, quantitative trait loci (QTL) affecting plant height have been identified in cereal crops such as rice (Li et al. 2003, Zhang et al. 2006b), maize (Zhang et al. 2006c) and wheat (Cui et al. 2011), as well as biomass crops such as sugarcane (Ming et al. 2002). Some molecular markers closely linked to QTL have even been developed for marker-assisted selection (Zhang et al. 2008), and some of dwarfing genes have been characterized or cloned (Ellis et al. 2005; Multani et al. 2003, Peng et al. 1999).
Plant height is also an important trait in upland cotton (Gossypium hirsutum L.), as this trait is closely related to canopy size and the photosynthetic capacity of the plant. Plant height primarily involves the number and length of the mainstem nodes and is determined by cell expansion during the growing season. Since the mainstem must support the necessary fruit branches and boll load by balancing vegetative and reproductive growth, appropriate plant height is essential for optimizing available sunlight and achieving maximum yield within a planting density. Although different growth environments and planting patterns allow for varying levels of individual plant height in cotton production (Zhang et al. 2006a; Reta-Sánchez and Fowler 2002), the global trend for machine picking makes shorter plants a better alternative, since taller plants are often associated with excessive vegetative growth and later maturity and can present harvesting difficulties (Percy et al. 2006). A reduction in plant height is usually achieved by frequently using plant growth regulators such as mepiquat chloride to control excessive vegetative growth (Siebert and Stewart 2006), which, if left unchecked, can lead to undesirable fruit shed, boll rot and subsequent yield reductions (Fowler and Ray 1977). However, successful breeding efforts in cereal crops clearly suggest the feasibility of plant architecture breeding to reduce plant height and to improve yield and fiber quality in cotton (Evans 1993; Gale et al. 1985).
Cotton plant height is inherited both qualitatively and quantitatively. There are many genes associated with plant height (Ellis et al. 2005). Several cotton dwarf mutants have been identified (Wu et al. 2009a; Harland 1918; Hutchinson and Ghose 1937), and some phytohormone signaling pathway-related dwarfing genes have been characterized (Liao et al. 2009; Aleman et al. 2008; Yang et al. 2006; Wilkins and Arpat 2005). Traditional quantitative genetic studies have revealed that plant height is also a complex trait, with additive effects (Wu et al. 2009b), both additive and dominant effects (Murtaza et al. 2006) and/or epistasis (Kalsy and Garg 1988; Khan and Khan 1993). Over the past decade, an increasing number of QTL for cotton plant height have been identified (Shappley et al. 1998; Wang et al. 2006; Saeed et al. 2011; Adawy et al. 2008; Song and Zhang 2009; Qin et al. 2009). However, the inconsistency of mapping results from these studies reveals the complicated multigenic characteristics of cotton plant height. In addition, few studies have involved analyses of digenic epistasis and QTL × environment (QE) interaction effects. Therefore, further studies employing different mapping populations and improved genetic maps are needed.
We previously identified QTL for plant architecture traits using an RIL population of Xiangzamian 2 (XZM2) cotton (Wang et al. 2006). In the present study, using an endensed genetic map mainly consisting of SSR markers, the same RIL population and an immortalized F2 (IF2) population (Wang et al. 2007; Liu et al. 2012, 2011) derived by random intercrosses between the RILs were used to identify main-effect QTL and epistatic QTL for cotton plant height. Results from this study will help elucidate the genetic basis of cotton plant height and provide useful markers for future breeding programs.
Materials and methods
Plant materials
The intraspecific cotton (G. hirsutum L.) hybrid XZM2 was developed from the cross “CRI12 × J8891” at the Hunan Cotton Research Institute, Changsha, China. The average plant height (PH) of this hybrid is between that of the parents. CRI12 is a pyramid-shaped cultivar with longer sympodial branches and shorter internode lengths, while J8891 is a column-shaped tall line with shorter sympodial branches and longer internode lengths. The parents used in this study were maintained by continued self-pollination. CRI12 was crossed with J8891 at Jiangpu Breeding Station, Nanjing Agricultural University (JBS/NAU) in 1998. F2 seeds were produced by selfing in the following winter in Hainan province. F2:3 seeds were produced in JBS/NAU in 1999 by selfing F2 plants derived from a single F1 plant. The development of the RIL population and the subsequent IF2 population is detailed in Liu et al. (2012). Briefly, A RIL population of 180 RIL families was constructed using a bulk-selfing technique. An IF2 population was made in 2003 in JBS/NAU by crossing between RILs randomly selected by two rounds of permutations. In each round of permutation, 180 RILs were randomly divided into two groups, where the lines were paired up at random, without replacement, to provide parents for 90 crosses. IF2 seeds were reproduced in 2007 by the same crosses as used above in JBS/NAU. The seeds produced from 180 crosses were used in the subsequent field trials.
Field trials and linkage map construction
The parents, the F1 population and 180 RILs were planted in Guanyun (34.33°N, 119.25°E), Jiangsu Province, China in 2003 and in JBS/NAU (32.04°N, 118.64°E) in 2007 in single-row plots. The parents, F1, and 171 IF2s were planted in JBS/NAU in 2004, 2005 and 2008 and in Linqing (36.86°N, 115.70°E), Shandong Province, China in 2008. The planting dates were from late March to early April in different years and at different locations. Seedlings up to 3–4 leaves were transplanted from seedbeds to fields, with 20 plants per row, at a plant-to-plant distance of 30 cm and a row-to-row distance of 80 cm. A randomized complete block design with two replications was used in all field trials. Ten representative plants in the middle of each row were tagged for measurement. The number of centimeters from the cotyledon nodes to the top of the main stem of each tagged plant was measured in September, roughly 2 months after topping. The mean values from the tagged plants were used for analysis. The molecular markers analyses and linkage maps construction were performed as described in Liu et al. (2012).
Data analysis
The mean of individual measurement from two replications in each environment was calculated. The difference between the two parents was detected by paired-samples t-tests. For QTL analysis, each site-year was analyzed separately as an individual environment. Further analysis was performed by combing two sites in 1 year and by combing all site-years within a population. Therefore, there were a total of three environments for the RILs and six environments for the IF2 population. Main-effect QTL analysis was conducted using Windows QTL Cartographer 2.5 (Basten et al. 2001) with the composite interval mapping (CIM) procedure (Zeng 1994). The standard model (Model 6), which takes forward stepwise regression with backward elimination, was adopted at a walk speed of 1 cM to search for QTL and to identify cofactors. Empirical significant LOD threshold values were estimated by 1,000 permutations (Churchill and Doerge 1994). A QTL was declared when the LOD score was greater than the threshold value. QTL confidence intervals (90 and 95 %) were set as map intervals corresponding to two and one LOD decline on either side of the peak. The degree of dominance of a QTL was estimated to be d/|a|.
Main-effect QTL tagging in the IF2 was also conducted using multimarker joint analysis (MJA) (Zhang and Xu 2005) and Windows QTL Cartographer 2.5 (Basten et al. 2001) with the multiple-interval mapping (MIM) procedure, respectively. The gene action modes were classified according to Stuber et al. (1987).
Digenic epistasis was evaluated using the MIM method of QTL Cartographer 2.5 (Basten et al. 2001), with the Bayesian information criteria (BIC-M0). QTL by environment interactions were tested by SAS v9.13 software (SAS Institute Inc., Cary, NC, USA) using MJA programs, along with multiple imputation techniques (100) (Liu et al. 2012). The penalized maximum likelihood method proposed by Zhang and Xu (2005) was used to estimate the parameters in the mixed linear model, and a detection rate of 0.30 was used to declare a significant QTL in MJA.
QTL nomenclature commonly used in rice was adopted (McCouch et al. 1997). In this nomenclature, the designation of a QTL begins with “q”, followed by an abbreviation of the trait name, the chromosome or linkage group and the serial number.
Results
Phenotypic variation in plant height of two immortalized populations
For plant height (PH), the parent J8891 demonstrated greater values than CRI12 in all environments examined, with significant differences between the two parents (Table 1). The values of the XZM2 (F1) plants were slightly lower than the mid-parent values in all environments, except for JBS/NAU 2005, in which the values of the F1 plants were similar to those of the higher-valued parent J8891.
The mean values of RILs and IF2 fell directly between those of the parents, with transgressive segregation in both directions. Some lines had even greater values than the taller parent J8891. The parents, F1 and IF2 had higher PHs in Linqing 2008 than in JBS/NAU 2004, 2005 and 2008, with PHs slightly shorter in JBS/NAU 2008 than in JBS/NAU 2004 and 2005. PH was negatively correlated with yield in the IF2; however, no significant correlation between PH and yield was detected in the RILs (data not shown). Trait values in the two populations fit normal distributions in all environments, with both skewness and kurtosis values less than 1.0. These results suggest that PH is eligible for QTL mapping.
QTL detected in the RILs and IF2
The RIL-based linkage map illustrated in Liu et al. (2012) was used to tag QTL for PH. A total of 19 QTL with genetic main effects and/or genetic × environment interaction effects were identified on 15 chromosomes or linkage groups by CIM, MIM and MJA in the RILs and IF2 populations; nine, seven and 14 were detected by CIM, MIM and MJA, respectively (Tables 2, 3; Fig. 1).

QTL for plant height detected by CIM, MIM and MJA in the RILs and IF2 derived from the cross of “CRI12 × J8891” (XZM2). Markers nearest to QTL detected in different environments are shown in red color. The digenic epistatic QTL pairs are shown in solid line. QTL detected solely by MJA had no confidence interval. For detailed genetic map, refer to Liu et al. (2012). (Color figure online)
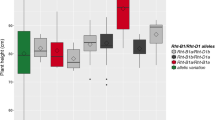
The partial dominant QTL qPH-D6-1 was detected by CIM, MIM and MJA in multiple environments in both the RILs and IF2, with LOD scores ranging from 3.13 to 8.50, which explained 6.86–20.04 % of the phenotypic variation (PV). The allele from CRI12 was responsible for a reduction in PH of 2.54 cm, and the dominant effects of this QTL reduced PH by 0.96 cm. The partial dominant QTL qPH-D8-2 was detected by CIM, MIM and MJA in the IF2, with LOD scores ranging between 3.90 and 8.60, explaining 9.63–20.32 % of the PV. A favorable allele from CRI12 reduced PH by 3.07 cm, and dominant effects of the QTL decreased PH by 1.16 cm. The partial dominant QTL qPH-D4-1 was identified by CIM in the RILs in two environments and by MJA in the IF2, explaining 6.10–13.72 % of the PV. A favorable allele from short parent CRI12 decreased PH by 1.91 cm, but the dominant effect of the QTL increased PH by 0.96 cm. Another partial dominant QTL, qPH-D11-1, was detected by CIM and MJA in the IF2, explaining 8.30 and 10.92 % of the PV, respectively, and the allele from CRI12 decreased PH by 2.54 cm, while the dominant effect was responsible for a reduction in PH of 0.71 cm. The additive QTL qPH-A5-1 was identified by CIM in the IF2, with a LOD score of 2.70, explaining 6.56 % of the PV. The allele from J8891 could reduce PH by 2.90 cm. The partial dominant QTL qPH-D9-2 was detected by MJA in the IF2, with a LOD score of 3.77 and a detection rate of 0.55. The beneficial allele from the short parent CRI12 decreased PH by 1.60 cm, compared with an increase of 0.81 cm by the dominant effect of the QTL. The partial dominant QTL qPH-A13-1 was detected by CIM in the IF2, with a LOD score of 2.91, explaining 8.69 % of the PV. The allele from J8891 could reduce PH by 2.75 cm, but the dominant effect of the QTL was associated with an increase in PH of 1.47 cm.
The other ten main-effect QTL detected by at least one of the three methods in the IF2 showed partial dominant to overdominant effects. The short parent CRI12 contributed alleles for decreasing PH at eight QTL, including qPH-D2-1, qPH-A4-1, qPH-A5-2, qPH-D5-1, qPH-D8-1, qPH-A9-1, qPH-D9-1 and qPH-LG01-1, but this parent contributed alleles for increasing PH at two QTL, qPH-D2-2 and qPH-A11-1, suggesting that favorable alleles for PH are distributed within the two parents. Dominant effects were associated with reducing PH for three QTL, including qPH-A5-2, qPH-D9-1 and qPH-LG01-1. In conclusion, additive effects of all 17 main-effect QTL collectively contributed to reducing PH by 31.22 cm over the population mean in homozygotes, and dominant effects of seven beneficial QTL were responsible for reducing PH by 9.18 cm in heterozygotes (Table 2).
In addition, five environment epistatic QTL were detected by MJA, with detection rates ranging from 0.30 to 0.90 and LOD scores between 3.46 and 6.56, each explaining 6.74–9.23 % of the PV (Table 3). The magnitude and even the direction of these QTL were different among environments, implying differential gene expression across environments. Three main-effect QTL, qPH-A5-1, qPH-D9-1 and qPH-A11-1, had significant QE interaction effects (Tables 2, 3). Since the main effects of qPH-A5-1 and qPH-D9-1 were detected by CIM and MIM, respectively, in the IF2, the detected a and d effects may have been confounded by QE interaction effects in the genetic model. Conversely, as qPH-A11-1 was detected only by MJA, its genetic effects should be expressed as the main effects (a and d) plus QE interaction effects at a specific environment. The other two QE interacting loci, qPH-A7-1 and qPH-A12-1, had no main effects (Table 3), suggesting that the expression of these two QTL is highly dependent on the environment.
Digenic epistasis detected by MIM in the IF2
Digenic epistasis comprises additive × additive (AA), additive × dominance (AD), dominance × additive (DA) and dominance × dominance interactions (DD). Five digenic epistatic QTL (E-QTL) pairs involving eight loci were detected for PH in different environments (Table 4; Fig. 1), jointly explaining 32.46 % of the PV. The interaction patterns were mainly AA and DD for all E-QTL pairs detected.
The QTL qPH-D8-1 near NAU3587 on D8 interacted with qPH-D8-2 near E21M8 on the same chromosome, with both AA and DD effects reducing PH by 1.09 and 4.34 cm, respectively, collectively explaining 8.85 % of the PV. The locus near TML05 on D10, which was not a significant main-effect QTL by itself, interacted with qPH-D5-1 near BNL3442 on D5 and qPH-D2-2 near CIR246 on D2. The combined DD effects of these loci could reduce PH by 7.39 and 6.47 cm, respectively, each explaining about 4.50 % of the PV. The other two E-QTL pairs were all involved in AA interactions. The QTL qPH-D6-1 interacted with the QTL qPH-A9-1 and qPH-D9-1, and each pair was associated with increased PH, explaining 9.92 and 4.63 % of the PV, respectively.
Discussion
In most cotton-growing regions, moderately shorter plants are frequently associated with an improvement in canopy structure and hence, population yield. Moderately short PH is a prerequisite for machine picking. Therefore, understanding the underlying genetic basis of cotton plant height is important to cotton architecture breeding programs. Dwarf mutants possessing dwarfing genes are usually too short to bear adequate fruit branches and boll load, and some of these mutants are even associated with sterility or other trait abnormalities (Wu et al. 2009a; Harland 1918; Hutchinson and Ghose 1937); therefore, their usage in cotton breeding is limited. Instead, semi-dwarf germplasms provide an ideal gene pool for genetic improvement of cotton. In this study, two high yielding cultivars CRI12 and J8891, which significantly distinguished by PH from each other, were used as parents for the construction of RILs and IF2 mapping populations, and the inheritance of PH was divided into QTL main effects, digenic epistasis and QTL by environment epistasis and examined using three mapping methods. The number of QTL detected by multi-environment-based MJA was much greater than that detected by CIM or MIM, demonstrating that the algorithm of MJA is a more powerful method for QTL detection.
Of the 17 main-effect QTL, one, nine, one and six QTL showed additive, incomplete dominant, dominant and overdominant effects, respectively. The additive effects of QTL with the CRI12 genotype could be positive or negative, which implies that QTL occurred in repulsion phase in the two parents. The additive effects of beneficial alleles from both parents could collectively reduce PH by 31.22 cm, suggesting the potential for pyramid selection. On the other hand, the net dominant effects of these QTL were associated with an increased PH of 9.54 cm, which is inconsistent with the observation that the PH in XZM2 was slightly lower than the mid-parent value in the majority of environments. These results suggest that the QTL for PH identified in this study are incomplete, as the genetic map only covered 20.20 % of the cotton genome (Liu et al. 2012). However, the results clearly demonstrate that both additive and dominant effects are important in the genetic constitution of PH, with additive effects playing a more important role in reducing PH. Analysis of variance was performed separately for the RIL and IF2 populations, which showed no significant G × E interactions (data not shown), whereas five QTL detected in the IF2 were involved in interactions with environment; three of these QTLs had significant genetic main effects, and two had no detectable main effects by themselves. The environment-dependent or modified expression of these QTL implies that QE interaction is also an important component affecting PH. These results are profoundly important to marker-assisted breeding, since the selection of QTL with significant QE interaction values may lead to unpredictable results in the progeny.
The main-effect QTL qPH-D2-1, qPH-D6-1, qPH-D8-2 and qPH-A9-1 were repeatedly detected across multiple environments and/or populations and showed less variation in direction and effects than the other QTL. In previous studies, Qin et al. (2009) identified five main-effect QTL for PH on D2, A5, D6, D9 and D10 using F2 and F2:3 populations derived from intraspecific crosses of “CR12 × J8891” and “CR12 × 4133”, of which three QTL (qPH-A5-1, qPH-D6-1 and qPH-D9-2) were detected in the present study characterized by the common neighboring markers of NAU879, CIR407 and BNL1672, respectively. We found that chromosomes A13 and D11 harbor QTL for PH, and similar results were also reported by Song and Zhang (2009), who used a BC1 population of an interspecific cross to identify QTL for plant architecture traits. These QTL should be particularly useful for marker-assisted manipulation of PH in upland cotton.
Digenic epistasis plays an important role in heredity and variation (Cheverud and Rountman 1995) and is regarded as the genetic basis of heterosis in crops (Yu et al. 1997; Hua et al. 2003; Melchinger et al. 2007; Kusterer et al. 2007; Shen et al. 2006). In this study, five digenic epistatic QTL pairs showing significant AA and/or DD effects were identified for PH. Seven out of eight interacting loci had significant genetic main effects, suggesting that these loci act as major genes as well as modifying genes in the expression of cotton plant height. Digenic epistasis should be considered in the utilization of QTL in cotton breeding, since the effect of a main-effect QTL involved in epistatic interactions is dependent on the genotypes of the other locus and can be negated by the genotypes of a second locus (Li et al. 2008). For example, the additive effects of alleles of CRI12 on QTL qPH-D6-1 significantly reduced PH (by 2.54 cm), but when this QTL interacted with qPH-A9-1 or qPH-D9-1, their AA effects increased PH by 2.20 and 3.64 cm, respectively. The percentage of loci with no genetic main effects involving digenic interactions was low compared with other reported values (Li et al. 1997). This is probably due to the MIM mapping procedure adopted in the present study, which is less powerful for the detection of epistasis between loci with small effects than other mapping procedures (Wang et al. 2005).
The QTL positions and effects detected in this study were not comparable to most other reported results due to the different markers used and the linkage groups unassigned (Shappley et al. 1998; Saeed et al. 2011; Adawy et al. 2008). However, with the improvements of map resolution and more mapping populations constructed via various crosses, more and more QTL will be disclosed through mapping efforts, which will contribute to our understanding of the genetic basis of cotton plant height.
References
Adawy SS, Diab AA, Atia MAM, Hussein EHA (2008) Construction of genetic linkage map showing chromosomal regions associated with some agronomic traits in cotton. Intl J Plant Breed 2(1):27–38
Aleman L, Kitamura J, Abdel-mageed H, Lee J, Sun Y, Nakajima M, Ueguchi-Tanaka M, Matsuoka M, Allen RD (2008) Functional analysis of cotton orthologs of GA signal transduction factors GID1 and SLR1. Plant Mol Biol 68:1–16
Basten CJ, Weir BS, Zeng ZB (2001) QTL Cartographer, Version 1.15. Department of Statistics, North Carolina State University, Raleigh, NC
Cheverud JM, Rountman EJ (1995) Epistasis and its contribution to genetic variance components. Genetics 139:1455–1461
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Cui F, Li J, Ding A, Zhao C, Wang L, Wang X, Li S, Bao Y, Li X, Feng D, Kong L, Wang H (2011) Conditional QTL mapping for plant height with respect to the length of the spike and internode in two mapping populations of wheat. Theor Appl Genet 122(8):517–1536
Ellis MH, Rebetzke GJ, Azanza F, Richards RA, Spielmeyer W (2005) Molecular mapping of gibberellin-responsive dwarfing genes in bread wheat. Theor Appl Genet 111:423–430
Evans LT (1993) Crop evolution, adaptation and yield. Cambridge University Press, Cambridge
Fowler JL, Ray LL (1977) Response of two cotton genotypes to five equidistant spacing patterns. Agron J 69:733–738
Gale MD, Youssefian S, Russell GE (1985) Dwarfing genes in wheat. Progress in plant breeding. Butterworths, London, pp 1–35
Harland SC (1918) On the genetics of crinkled dwarf rogues in Sea Island cotton. West Ind Bull 16(1):82
Hua JP, Xing YZ, Wu WR, Xu CG, Sun XL, Zhang QF (2003) Single-locus heterotic effects and dominance-by-dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 100(5):2574–2579
Hutchinson JB, Ghose RLM (1937) On the occurrence of”crinkled dwarf” in Gossypium hirsutum L. J Genet 34(3):437–446
Kalsy HS, Garg HR (1988) Analysis of generation means for metric traits in upland cotton (Gossypium hirsutum L.). Indian J Agric Sci 58:397–399
Khan MNU, Khan IA (1993) Study of gene action for some morphological plant characters in upland cotton. Pak J Agric Sci 30:94–98
Kusterer B, Muminovic J, Utz HF, Piepho HP, Barth S, Heckenberger M, Meyer RC, Altmann T, Melchinger AE (2007) Analysis of a triple testcross design with recombinant inbred lines reveals a significant role of epistasis in heterosis for biomass-related traits in Arabidopsis. Genetics 175:2009–2017
Li ZK, Pinson SRM, Park WD, Paterson AH, Stansel JW (1997) Epistasis for three grain yield components in rice (Oryza sativa L.). Genetics 145:453–465
Li ZK, Yu SB, Lafitte HR, Huang N, Courtois B, Hittalmani S, Vijayakumar CHM, Liu GF, Wang GC, Shashidhar HE, Zhuang JY, Zheng KL, Singh VP, Sidhu JS, Srivantaneeyakul S, Khush GS (2003) QTL × environment interactions in rice. I. Heading date and plant height. Theor Appl Genet 108:141–153
Li LZ, Lu KY, Chen ZM, Mu TM, Hu ZL, Li XQ (2008) Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics 180:1725–1742
Liao WB, Ruan MB, Cui BM, Xu NF, Lu JJ, Peng M (2009) Isolation and characterization of a GAI/RGA-like gene from Gossypium hirsutum. Plant Growth Regul 58:35–45
Liu RZ, Wang BH, Guo WZ, Wang LG, Zhang TZ (2011) Differential gene expression and associated QTL mapping for cotton yield based on a cDNA-AFLP transcriptome map in an immortalized F2. Theor Appl Genet 123:439–454
Liu RZ, Wang BH, Guo WZ, Qin YS, Wang LG, Zhang YM, Zhang TZ (2012) Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid in Gossypium hirsutum L. Mol Breeding 29:297–311
McCouch SR, Cho YG, Yano PE, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newslett 14:11–13
Melchinger AE, Piepho HP, Utz HF, Muminovic J, Wegenast T, Törjék O, Altmann T, Kusterer B (2007) Genetic basis of heterosis for growth-related traits in Arabidopsis investigated by testcross progenies of near-isogenic lines reveals a significant role of epistasis. Genetics 177(3):1827–1837
Ming R, Del Monte TA, Hernandez E, Moore PH, Irvine JE, Paterson AH (2002) Comparative analysis of QTLs affecting plant height and flowering among closely-related diploid and polyploid genomes. Genome 45:794–803
Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS (2003) Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302:81–84
Murtaza N, Qayyum A, Malik W, Noor E (2006) Genetic study of yield of seed cotton and plant height in cotton genotypes. Int J Agri Biol 8(5):630–635
Peng J, Richard DE, Hartley NM, Murphy PG, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP (1999) “Green revolution” genes encode mutant gibberellin response modulators. Nature 400:256–261
Percy RG, Cantrell RG, Zhang J (2006) Genetic variation for agronomic and fiber properties in an introgressed recombinant inbred population of cotton. Crop Sci 46:1311–1317
Qin YS, Liu RZ, Mei HX, Zhang TZ, Guo WZ (2009) QTL mapping for yield traits in Upland cotton (Gossypium hirsutum L.). Acta Agronomica Sinica 35(10):1812–1821
Reta-Sánchez DG, Fowler JL (2002) Canopy light environment and yield of narrow-row cotton as affected by canopy architecture. Agron J 94:1317–1323
Saeed M, Guo WZ, Ullah I, Tabbasam N, Zafar Y, Rahman M, Zhang TZ (2011) QTL mapping for physiology, yield and plant architecture traits in cotton (Gossypium hirsutum L.) grown under well-watered versus water-stress conditions. Electron J Biotechnol, 14(3). 10.2225/vol14-issue3-fulltext-3
Shappley ZW, Jenkins JN, Zhu McCarty JC (1998) Quantitative traits loci associated with agronomic and fiber traits of upland cotton. J Cotton Sci 4:153–163
Shen XL, Zhang TZ, Guo WZ, Zhu XF, Zhang XY (2006) Mapping fiber and yield QTLs with main, epistatic, and QTL × environment interaction effects in recombinant inbred lines of cotton. Crop Sci 46:61–66
Siebert JD, Stewart AM (2006) Influence of plant density on cotton response to mepiquat chloride application. Agron J 98:1634–1639
Song XL, Zhang TZ (2009) Quantitative trait loci controlling plant architectural traits in cotton. Plant Sci 177:317–323
Stuber CW, Edwards MD, Wendel JF (1987) Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Sci 27:639–648
Wang H, Zhang YM, Li XM, Masinde GL, Mohan S, Baylink DJ, Xu SZ (2005) Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170:465–480
Wang BH, Wu YT, Huang NT, Zhu XF, Guo WZ, Zhang TZ (2006) QTL mapping for plant architecture traits in Upland cotton using RILs and SSR Markers. Acta Genetica Sinica 33(2):161–170
Wang BH, Wu YT, Guo WZ, Zhu XF, Huang NT, Zhang TZ (2007) QTL analysis and epistasis effects dissection of fiber qualities in an elite cotton hybrid grown in second-generation. Crop Sci 47:1384–1392
Wilkins TA, Arpat AB (2005) The cotton fiber transcriptome. Physiol Plant 124:295–300
Wu CT, Zhou BL, Zhang TZ (2009a) Isolation and characterization of a sterile-dwarf mutant in Asian cotton (Gossypium arboreum L.). J Genet Genomics 36(6):343–353
Wu JX, McCarty JC, Saha S, Jenkins JN, Hayes R (2009b) Genetic changes in plant growth and their associations with chromosomes from Gossypium barbadense L. in G. hirsutum L. Genetica 137:57–66
Yang SS, Cheung F, Lee JJ, Ha M, Wei NE, Sze S-H, Stelly DM, Thaxton P, Triplett B, Town CD, Chen ZJ (2006) Accumulation of genome-specific transcripts transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. Plant J 47:761–775
Yu SB, Li JX, Tan YF, Gao YJ, Li XH, Zhang QF, Saghai Maroof MA (1997) Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 94:9226–9231
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang YM, Xu S (2005) A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity 95:96–104
Zhang PT, Zhu XF, Guo WZ, Yu JZ, Zhang TZ (2006a) Inheritance and QTLs tagging for ideal plant architecture of Simian 3 using molecular markers. Cotton Sci 18:13–18
Zhang YS, Luo LJ, Xu CG, Zhang QF, Xing YZ (2006b) Quantitative trait loci for panicle size, heading date and plant height co-segregating in trait-performance derived near-isogenic lines of rice (Oryza sativa). Theor Appl Genet 113:361–368
Zhang ZM, Zhao MJ, Ding HP, Rong TZ, Pan GT (2006c) QTL mapping analysis of plant height and ear height of maize (Zea mays L.). Genetika 42(3):391–396
Zhang KP, Tian JC, Zhao L, Wang SS (2008) Mapping QTLs with epistatic effects and QTL × environment interactions for plant height using a doubled haploid population in cultivated wheat. J Genet Genomics 35:119–127
Acknowledgments
This work was financially supported in part by the 12th Five-Year National Science and Technology Support Program (2011BAD35B05-1) and Shandong Agricultural Bioresources Innovation and Utilization Program—“Mining of Cotton Functional Genes and Elite Germplasm Improvement”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, R., Ai, N., Zhu, X. et al. Genetic analysis of plant height using two immortalized populations of “CRI12 × J8891” in Gossypium hirsutum L.. Euphytica 196, 51–61 (2014). https://doi.org/10.1007/s10681-013-1013-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-013-1013-0