
Abstract
Genetic diversity analysis within a species is vital for understanding evolutionary processes at the population and genomic levels. We report a detailed study of molecular diversity, polymorphism and linkage disequilibrium in three groups of rice (Oryza) germplasm accessions based on 176 SSR markers. The first group included 65 rice (O. sativa L.) accessions introduced from seven countries, including five regions of China. The second group included 58 US rice varieties released in the past 25 years. The third group consisted of 54 accessions of rice wild relatives represented by ten different species. The number of alleles per SSR marker ranged from 4 to 32 with a mean of 16 alleles and the polymorphism information content values ranged from 0.43 to 0.91 with a mean of 0.70. The variation in SSR alleles was a significant contribution to the genetic discrimination of the 177 accessions within the three Oryza groups. Analysis of molecular variance identified deviation from Hardy–Weinberg equilibrium. Principal coordinates analysis clearly separated the accessions into their respective three groups. Neighbor-joining phylogenetic cluster reflects the ordination of each accession. Linkage disequilibrium (D′) averaged 0.75 in wild Oryza spp., and about 0.5 in both US and international O. sativa accessions. Our results showed that LD among adjacent loci in both O. sativa and Oryza spp. accessions is strong enough to be detecting marker-trait association via genome-wide scans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic mapping in plants up to now involves the development, genotyping and phenotyping of a random sample of lines derived from a single cross of two accessions. This type of mapping population shows extreme disequilibrium between loci (Mather et al. 2004), and is used to detect quantitative trait loci (QTL) for phenotypic traits and to map these traits to an approximate location in the genome. Both QTL and association mapping approaches require LD between markers and genes underlying a phenotypic trait. In contrast to QTL studies, where LD is generated by the mating design, association methods rely entirely on the LD present in the germplasm under study, therefore chromosome segments in LD are a prerequisite for association methods (Maurer et al. 2006). Ideally these segments contain a marker and a locus responsible for the trait of interest. Association studies can be carried out by testing for association of phenotypic traits with markers spread across the entire genome (genome scan approach) or with markers in regions of candidate genes (candidate gene approach). By contrast, genetic mapping of humans relies on analysis of genotypic and phenotypic data sampled in a non-random fashion from existing populations of complex but often unknown structure (Kruglyak 1999).
The non-random association of alleles at different loci is commonly referred to as linkage disequilibrium (LD) and has become an important aspect of studies on population structure in animals, plants and human evolution (Sawyer et al. 2005). In such populations, LD may decline within relatively short distances in the genome, which makes fine mapping possible. A variety of statistics have been used to measure LD for different purposes with the two most common being r 2 and D′ (Flint-Garcia et al. 2003). For LD between two multi-allelic loci, D′ was applied in Oryza (Thomson et al. 2007), Triticum (Maccaferri et al. 2005) and Zea mays L. (Maurer et al. 2006; Stich et al. 2005). Multi-allelic markers like SSR markers were used in LD-based association studies in Triticum (Kruger et al. 2004; Maccaferri et al. 2005), Hordeum vulgare L. (Kraakman et al. 2004, 2006), Z. mays (Stich et al. 2005) and Lolium perenne L. (Skøt et al. 2005) to identify the position of the genes conferring a specific trait or phenotype. In sugar beet (Beta vulgaris L.), LD extended up to three cM (Kraft et al. 2000), while LD in some Arabidopsis populations exceeds 50 cM (Nordborg et al. 2002). LD as a function of genetic distance is very common for distances <10 cM (Kraakman et al. 2004) in barley (H. vulgare), a self-pollinating species, as compared to Z. mays, an out-crossing species, where the LD declined at distance >2000 bp (Remington et al. 2001). Population structure and its effect on diversity and LD surrounding the Xa5 locus using 21 Simple sequence repeat (SSR) from 114 rice accessions was reported (Garris et al. 2003). These populations were highly structured and significant LD was observed between sites up to 100 kb apart. This is comparable to the level of LD in flowering time gene FRIGIDA in A. thaliana (Johanson et al. 2000) but differs markedly from that in dwarf8 and other genes in maize (Thornsberry et al. 2001). In another study of Arabidopsis, the LD decayed rapidly within 50 kb (Nordborg et al. 2005) as compared to one human population where the LD extended over 3 cM (Eaves et al. 1998).
SSR markers have been widely used in rice (Oryza sativa L.) germplasm evaluation for both international (Ishii and McCouch 2000; Ishii et al. 2001; Semon et al. 2005; Thomson et al. 2007) and domestic US (Ni et al. 2002; Lu et al. 2005) collections. These studies also revealed the classification within the Asian cultivated rice, O. sativa, into indica and japonica based on their gross morphology and growth habitats (Chang 1995) and identification of the wild species O. rufipogon Griff. and O. nivara Sharma et Shastry as the ancestors of O. sativa. The use of SSRs to interpret population structure provides much greater resolution than other types of markers because of the high level of polymorphism at SSR loci (Akkaya et al. 1992; Cho et al. 2000). In rice, the highly polymorphic nature of SSR motifs is coupled with a low level of homoplasy observed in O. sativa cultivars (Chen et al. 2002), providing an appropriate tool for population genetic studies (Lu et al. 2005). The number of markers needed for a genome-wide association mapping depends on the level of LD extends in the populations of interest. LD mapping could complement and enhance previous QTL information for marker-assisted selection in wheat (Breseghello and Sorrells 2006), barley (Kraakman et al. 2006) and maize (Yu and Buckler 2006). LD is considered especially valuable in studies to map genes associated with common complex traits. These traits have very low mutation rates, thus, nonrandom association between different allelic forms must be defined.
Frequent disequilibrium among non-linked SSR markers within diverse sets of barley germplasm accessions was observed, suggesting that association mapping without consideration of population structure would have a high Type I error rate (Mather et al. 2004). With sufficient marker density and control of population structure, patterns of disequilibrium among loci may be appropriate for association mapping of trait loci. However, LD patterns were found to vary substantially between human among populations (Sawyer et al. 2005). The significant quantitative and qualitative variation in LD among populations, even for populations within a geographic region, indicates the importance of studying diverse populations in any project to assure broad applicability of the results. The objectives of this study were to analyze the molecular genetic diversity in a diverse collection of rice germplasm and estimate the pairwise LD between SSR markers that covered the complete rice genome in three groups of Oryza accessions, O. sativa from international sources and the US and wild Oryza spp.
Materials and methods
The background of the three groups of rice accessions used in this study is given in Table 1. The first group included 65 accessions introduced from seven countries, including five regions of China (Eizenga et al. 2006). The second group included 58 US varieties released in the past 25 years or used as parents in US rice breeding programs (Lu et al. 2005). The third group consisted of 54 rice wild relatives (Oryza spp.) represented by ten different species (Eizenga et al. 2007).
Seedling growth and DNA extractions for the US varieties were according to Lu et al. (2005). Seedling germination and DNA extraction for the O. sativa accessions from international sources and the wild Oryza spp. were according to Eizenga et al. (2006) with the wild Oryza spp. accessions going through a tissue culture step for good germination (Eizenga et al. 2007). One hundred and seventy-six markers located on the twelve chromosomes were selected from the SSR set developed and mapped by McCouch et al. (2002). The SSR covered a total of 1745.5 cM of rice genome with an average of 10.6 cM between adjacent loci. DNA amplifications were performed using an MJ Research PTC-100 96 Plus thermal cycler. PCR products were size-separated using an ABI Prism 3700 DNA analyzer (Applied Biosystems, Foster City CA). SSR fragment sizing was performed with GenScan® 3.1.2 software (Applied Biosystems, Foster City, CA) using the default analysis settings. Alleles were called with Genotyper® 2.5 (Applied Biosystems, Foster City, CA), and binned manually.
PowerMarker software (Liu and Muse 2005) was used to calculate gene diversity, allele frequencies and PIC values (Botstein et al. 1980) for each SSR marker. F IT , deviations from Hardy–Weinberg expectation across the whole population; F IS , deviation from Hardy–Weinberg expectation within a population; and F ST , correlation of alleles within subpopulations, were calculated using an analysis of molecular variance (AMOVA) approach (Weir 1996; Schneider and Excoffier 1999) implemented in Arlequin ver 3.11 (Excoffier et al. 2005). Principal coordinates analysis (PCA) of the worldwide Oryza accessions was performed based on Nei (1973) distance matrix using GenAlEx 6 (Peakall and Smouse 2006). Genetic distances between the 177 accessions were evaluated according to Nei (1973) with the phylogenetic tree being built by neighbor-joining (Saitou 1987). D′ was calculated for all pairs among the 176 SSRs according to Tenesa et al. (2003) using PowerMarker (Liu and Muse 2005). The extent of pairwise LD among marker loci pairs was estimated as a function of the distance among SSR markers.
Results
Allele diversity
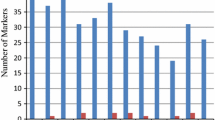
Over all the accessions, a total of 1616 alleles were detected for all 176 SSRs. The average number of alleles ranged from 4 to 32 and was in average of 15.8 (Table 2, Fig. 1a). Within the O. sativa from international source, the number of alleles ranged from 2 to 18 with a mean of 7.6. In US cultivars, the number of alleles per locus ranged from 2 to 15 with an average of 4.5 where in Oryza spp., the number of alleles ranged from 3 to 26 with an average of 12.4.

Distribution of the (a) number of alleles and (b) PIC values detected with 176 SSR markers in the 177 rice accessions that were genotyped and within the three groups [65 O. sativa from international sources, 58 US cultivars and 54 wild relatives (Oryza spp.)]
The relative informativeness of each marker can be evaluated on the basis of its PIC value (Table 2, Fig. 1b). The PIC values ranged from 0.43 to 0.91 with a mean of 0.73. The PIC values of each single SSR are reflected in the gene diversity computed according to Nei (1973), which varied from 0.15 (RM5752, chromosome 7) to 0.92 (RM333, chromosome 10), with the average of 0.73 (Table 2). The wild Oryza species have the highest PIC value (0.78) while the US cultivars revealed the lowest value (0.39). Again the O. sativa obtained from various regions had a higher PIC value of (0.57) than the US cultivars but not as high as the Oryza spp. group. For the US accessions, gene diversity averaged 0.43, ranging for the single marker from 0.03 to 0.86 whereas the accessions from international sources displayed higher values for gene diversity ranging from 0.16 to 0.89. The gene diversity was significantly higher in the wild Oryza spp. accessions (0.80) than the international and US O. sativa accessions.
Molecular genetic variance
The distribution of molecular genetic variation among and within the three populations of accessions was estimated by analysis of molecular variance, AMOVA (Table 3). AMOVA revealed that 32% of the total variation was among the three groups, while 68% was among individuals within the groups. Calculation of Wright’s F statistic at all SSR loci revealed that F IS was 0.72 and F IT was 0.79. Determination of F ST for the polymorphic SSR loci across all accessions revealed F ST ranged from 0.08 for marker RM122 to 0.75 for RM507 on chromosome 5 with an average of 0.33. Determination of the pairwise F ST values between the three Oryza populations (Table 4) indicated the genetic differentiation was the highest for the combination of wild Oryza spp. and US accessions (F ST = 0.77), and wild Oryza spp. and international (F ST = 0.62). The F ST value for international and US accessions was 0.16.
Genetic distance between the accessions
PCA was used to characterize substructure of the three groups of rice accessions and to account for the inbreeding nature of rice. A 2-dimensional scatter plot involving all 177 rice accessions revealed that the two PCA axes accounted for 48.6% and 20.1% of the genetic variation among populations (Fig. 2). The analysis revealed distinct clustering for accessions from the three different backgrounds. However, the only accessions originating from the USA were clearly separated from the other accessions. The US accession imported from China, TeQing (no. 120), was separated from the other US accessions on the first and second axis by its strongly positive and negative eigenvector (0.827, −0.501, respectively) as opposed to the negative values for the other US accessions. The accessions from international sources formed a single discrete cluster while most accessions from USA also formed a cluster. In contrast, the wild Oryza spp. accessions clustered in a wide region between international and US accessions.

Principal coordinates analysis (PCA) of 177 rice accessions [65 O. sativa from international sources (Int), 58 US cultivars and 54 wild relatives (Oryza spp.)] using 176 SSR markers based on Nei (1973) distance estimates. PC1 and PC2 are the first and the second principal coordinates, respectively. Numbers in parentheses refer to the proportion of variance explained by the principal coordinate
Scattergrams of the first two axes of a PCA for the three groups of accessions are presented in Fig. 3. The first two PCA axes explain 46.1 and 20.7% of the genetic variation among the international accessions, respectively (Fig. 3a). A group of seven accessions [Akitakomachi (no. 8), GZ-5578-2-1-2 (no. 15), GZ-5830-48-2-2 (no. 16), Iac_47 (no. 17), Nipponbare (no. 26), Wab450-24-3-2-P18-hb (no. 58), Wab450-I-B-P-62-hb (no. 59)] are strongly separated and concentrated in the positive axes. Accessions Koshihikari (no. 24), Milyang (no. 25) and Pyongyang_23 (no. 28) are intermediate in the PCA.

Scattergram of the first two axes of a Principal coordinates analysis (PCA) of the worldwide Oryza accessions; (a) international accessions, (b) US cultivars, and (c) wild relatives Oryza spp. Numbers in the plots represent the accessions listed in Table 1
The PCA of US rice is shown in Fig. 3b where axis PC1 explained 45.5% of the variation and axis PC2 15.7% of the variation. A group of temperate japonica cultivars from California (nos. 74, 75, 97, 98, 99, 100, 102, 115 and 116) was clustered in the PCA separated from the tropical japonica cultivars both medium grain (nos. 93, etc.) and long grain (90, etc.) types grown in the Southern USA. Figure 3c depicts a two-dimensional scatterplot involving 54 wild rice relatives (Oryza spp.). The PCA axes accounted for 25.1 and 23.1% of the total variance. The accessions most clearly separated were the African cultivated rice O. glaberrima (nos. 134, 135, 136) and its ancestral parent O. barthii (nos. 128, 129, 130, 131, 132), from the ancestral parents, O. nivara and O. rufipogon, of cultivated rice, O. sativa. Cluster analysis of the genetic distance matrix developed by neighbor-joining method (Fig. 4) resulted in a phenetic tree that reflected ordination consequences with each accession and identified the three major groups.

Neighbor-joining phylogenetic tree for 177 rice accessions based on Nei (1973) genetic distance estimates. The three major groups and sub-clusters are identified
Extent of linkage disequilibrium (LD)
The distribution of D′ values (Fig. 5) from linked SSR markers in the international accessions and US cultivars ranged from 0.0 to 1.0 with an average of 0.540 and 0.595, respectively. Extensive LD in US rice was observed among linked and unlinked SSR (Fig. 5b). The values of D′ in the wild Oryza spp. group ranged from 0.244 for markers to 1.0 with an average of 0.752. The plot of LD across the wild accessions (Fig. 5c) revealed that the association across the entire genome was very high whereas the extent of LD across the international accessions (Fig. 5a) was similar to that observed across the US cultivars (Fig. 5b). Although the overall trend reveals a gradual decrease of LD with physical distance, there was an undulating pattern in the levels of association among the different between-marker comparison groups in all three-sample groups studied.

Distribution of D′ values observed between linked SSR marker pairs as a genetic distance in centiMorgans (cM) for each individual group of rice accessions; (a) international O. sativa, (b) US O. sativa cultivars and (c) wild Oryza spp. The LD between unlinked loci are presented at 250 cM
Discussion
Allele diversity
The narrow germplasm base of the US is reflected in low number of alleles especially when compared with the O. sativa accessions from around the world and the even wider variation in the Oryza spp. accessions. Calculation of Wright’s F statistic at all SSR loci revealed that there was a deviation from Hardy–Weinberg expectation for molecular variation within the populations, and the high F IT indicatined non-equilibrium conditions across the three groups and a lack of heterozygosity. This lack of equilibrium is most likely due to rice being an inbred species. F ST for the polymorphic SSR loci across all accessions revealed 33.1% of the total variation in allele frequency of the 177 accessions was due to genetic differences among populations. The F ST value for international and US accessions was the smallest. These values confirm that the US accessions were quite different from most other O. sativa accessions.
PIC values observed by Xu et al. (2005) for 60 SSR markers in the world collection ranged from 0.17 to 0.92 with an average of 0.74 and in the US collection ranged from 0.02 to 0.88 with an average of 0.50. Even though the PIC values calculated by Xu et al. (2005) are higher than those calculated in this study, the group containing US cultivars has a lower PIC value than the group from international sources. The higher PIC value for the world collection (Xu et al. 2005) probably is due to the accessions being chosen from international sources, whereas the O. sativa accessions in this study initially were selected as blast-resistant in field tests. A collection of 111 lines from 22 countries studied by Xu et al. (2005) had a range of 2–34 alleles with accessions having five or six alleles being the most frequent. This can be compared to the 145 US rice accessions in a previous study (Lu et al. 2005) where the most frequent number of alleles was four with the number of alleles ranging from 2 to 20. In a genetic diversity study by Garris et al. (2005), the indica and tropical japonica groups contained a high percentage of polymorphic loci (99%) and an average of 7.26 and 6.09 alleles per locus, respectively. The temperate japonica group had lower diversity with 91% polymorphic loci and 4.9 alleles per locus. Also, the average PIC value (0.67) across cultivars was high, similar to the PIC value (0.70) in this study.
Genetic distances among the populations
The genetic diversity between the individuals subjected to an association study can affect the detection of marker-trait associations. If individuals from subpopulations differing in allele frequencies are analyzed together, assuming only one base population, then LD between linked as well as between unlinked loci will be detected. Successful mapping of genes however, requires strong LD only between markers that are tightly linked to the loci underlying the trait of interest. As a consequence, association studies in admixtures of subpopulations with different allele frequencies are expected to suffer from a high rate of false positives (Maurer et al. 2006). A PCA plot can be regarded as a tool for visualizing the differences in allele frequencies of the individual accessions. Subsequently, a PCA plot can be used to check visually for a substructure within the sample of individuals thus, avoiding a high rate of false positives due to population admixture in an association study. The separation of international and US accessions in the PCA plot was in accordance with earlier studies (Eizenga et al. 2006; Agrama et al. 2007). PCA plots for the individual pools did reveal further subgrouping within sets. The results of the F ST analysis (Wright 1978) indicated moderately large differences between populations. Our results showed that association mapping could be carried out on a combination of the three groups, depends on the used statistical methods, to identify the QTL of interest. In contrary, in a study on Zea mays, the results from the diversity analysis suggest carrying out association studies separately within pools (Maurer et al. 2006).
Cluster analysis revealed that group 1 consisted of the majority of the international accessions; group 2 was all the US rice cultivars, while group 3 included most of the wild Oryza spp. The international O. sativa accessions in group 1 had two sub-clusters, one originating from China and the other from Africa. A small group of the international O. sativa clustered close to the US cultivars (Eizenga et al. 2006) possibly due to being a japonica type whereas the majority of the accessions were the indica type as previously described. The US O. sativa cultivars, group 2, were the least divergent, all clustering together with sub-clusters of temperate japonica from California, tropical japonicas with medium grain type and tropical japonicas with a long grain type (Lu et al. 2005). The wild Oryza spp., group 3, were the most divergent and sub-clustered roughly into (a) the African cultivated rice, O. glaberrima, and its ancestral progenitor, O. barthii and (b) the ancestral progenitors of O. sativa, both O. nivara and O. rufipogon as discussed further in Eizenga et al. (2007).
The largest and most geographically distributed group of wild Oryza spp. germplasm embodied more genetic diversity than either international O. sativa accessions (Group 1) or US cultivars (Group 2). The three groups also differ for several population genetic statistics that offer insight into their respective genetic background. In all analyses using RAPD, RFLP, nuclear SSLP and chloroplast SSLP (Bautista et al. 2001), O. sativa and O. glaberrima clustered with their respective ancestral parents, O. rufipogon or O. nivara, and O. barthii. However the type of clustering with other wild species was different. The results of RAPD and RFLP analyses indicate that O. glumaepatula was relatively close to the groups of O. sativa and O. glaberrima whereas O. longistaminata A. Chev. et Roehr. and O. meridionalis Ng were highly differentiated from other A-genome species.
Linkage disequilibrium patterns
In QTL mapping studies, the LD required for gene mapping is generated as a result of the mating design, while association studies exploit the LD present in the population of interest (Maurer et al. 2006). The distance between SSR markers currently employed for routine fingerprinting in plant breeding programs typically ranges over several cM (Agrama et al. 2007). In large populations of autogamous species, LD is expected not to decline with each reproductive cycle and stretches of LD extending over several cM are expected. Consequently, association mapping with SSRs in such populations does seem very promising. To represent LD, D′ was assessed as a function of the inter-marker distance for the three individual groups. D′ decreased most rapidly in the international group whereas the group of wild relatives showed extensive variability. These results provide strong evidence that long-range LD exists in wild relatives. This could be a result of strong selection of particular allele limits genetic diversity around a locus, resulting in increases of LD around the selected gene. It also, gene-gene or gene-environment can differ between populations, for instant, if a particular genetic variation were only manifest in populations with a particular environmental background (Hirschhorn et al. 2002). Higher LD in wild accession could, also, be a result of population stratification.
Population structure can result from geographic isolation factors (for example, isolation-by-distance phenomenon). Association results of SSR pairs, especially those less than 25 cM apart, identified the presence of an extensive LD within each group. Similar levels of long range LD, extending over several cM, have been reported for self-pollinating species such as Arabidopsis when considering isolated populations (Nordborg et al. 2002), barley (Russell et al. 2003), soybean (Hyten et al. 2007) and wheat (Maccaferri et al. 2005). As spurious associations between phenotypes and marker loci can be caused by population structure, the extent and structure of LD within a sample population must be known before selecting an appropriate association mapping strategy (Lander and Schork 1994). This is particularly true when inbreeding crop species such as rice and barley are examined where these complicating factors are expected to be more prevalent (Rostoks et al. 2006). Rice, in addition to being an inbreeder, has undergone a severe population bottleneck during domestication (Tanksley and McCouch 1997). In contrast, low LD was identified in wild barley despite a high rate of self-fertilization (Morrell et al. 2005). LD between adjacent loci in rice breeding programs is strong enough to be useful in marker-trait association studies via genome-wide scans that exploit LD to map common multigenic traits such as grain yield and quality. The pattern of LD in several US accessions has shown extensive LD with a decaying on a genome-wide scale over 50–100 cM. The founder effect and population bottlenecks are factors in which LD in a young or recovering population has not had time to decay through recombination, self-fertilization and inbreeding. Indeed, the short breeding history of US rice (Lu et al. 2005), it is possible that the decay of LD observed in these populations is mainly due to outcrossing and recombination events that have taken place in the last 100 years. The same observation has been identified in US Arabidopsis populations, which has introduced to North America 200 years ago (Nordborg et al. 2002).
Under recurrent self-fertilization, the level of heterozygosity decays at the rate of one-half per locus per generation, thus within a few generations, a self-fertilizing population is expected to consist entirely of homozygous lines. Consequently, there is an extreme reduction in the rate of effective recombination and the decay of LD will be arrested. Generally this been observed in studies of natural populations where within-population levels of LD for isozyme polymorphisms were generally higher in populations of self-fertilizing plants than in outcrossing plants (Brown 1979; Hastings 1990). A number of factors can contribute to excess intralocus LD, including selection, species-wide reductions in effective population size, population stratification, or geographic structure (Morrell et al. 2005). The large LD observed for close markers in the wild Oryza spp. population as compared to the cultivated O. sativa population, is strong evidence for the wild Oryza spp. being the ancestral species. This phenomenon has been observed in other species such as barley (Rostoks et al. 2006). Many plant species have a mixed mating system with admixture of outcrossing and self-fertilizing, where occasional outcross events produce new heterozygous lines that within a few generations sort out into homozygous lines. Under this scenario, LD decays at a rate that is a function not only of recombination distance but also the level of outcrossing (Morrell et al. 2005).
In our experimental data, the average D′ of linked loci in LD ranged from 0.54 in US O. sativa accessions to 0.75 in wild germplasm. Regarding the prospects of association tests, this observation implies that: (1) the high percentage of loci pairs in LD in the wild Oryza spp. group should allow association tests to successfully map genes providing the population size is large enough so that the tests have sufficient power and (2) the LD present in the O. sativa group though smaller was similar to the wild Oryza spp., therefore comparable rates of LD between loci are expected, given a sufficiently large sample size. Genome-wide association studies depend on the extent of LD, the number and distribution of markers, and the underlying structure in the populations being evaluated. Outbreeding species generally show limited LD, consequently more markers are required for effective whole-genome association genetic scans. In contrast, several of the world’s major food crops are self-fertilizing inbreeding species with narrow genetic bases and theoretically extensive LD. DNA sequencing in the two genomic regions of Oryza showed that levels of LD are remarkably different between O. rufipogon and O. sativa ssp. indica. LD in O. rufipogon decays within 5 kb, whereas it extends to 50 kb in O. sativa ssp. indica (Rakshit et al. 2007). This difference in the extent of LD may partly be explained by the breeding behavior of the species. Lower levels of LD in O. rufipogon are expected because O. rufipogon experiences a higher rate of outcrossing than O. sativa, which is a strictly self-pollinated crop (Khush 1997). LD decays rapidly in maize, a cross-pollinated crop, making this phenomenon an ideal tool for association studies. In maize landraces and a broad sample of tropical and temperate inbreds, LD often declines to nominal levels within 1.5 kb, whereas elite breeding material has less rapid decay (Buckler et al. 2006). However in Arabidopsis, a selfing species, LD decays within approximately 1 cM, or 250 kb to 20 kb based upon global or local populations as a result of founder events (Nordborg et al. 2002, 2005). For practical purposes, the low LD in O. rufipogon would be advantageous for association-based cloning of genes controlling phenotypic traits by using a large number of markers. In O. sativa ssp. indica, we will be able to use markers spaced 50 kb apart on average to carry out a rough agronomic trait mapping and marker-assisted selection.
Conclusion
This study suggests that a relatively small number of SSR evenly spaced along the genetic linkage map, could be sufficient for an initial whole-genome association scan in inbreeding crop plants, such as rice, in an appropriate set of germplasm. Whether such numbers will be sufficient for detecting marker associations with complex traits needs to be experimentally determined. In addition to examine the suitability of populations for association mapping experimentally there exist, the possibility of performing computer simulations. Although association scans on such a scale would generally not result in the identification of a small set of candidate genes for a given trait, the observed extent of LD would ensure resolution relevant for existing breeding programs and provide a rational basis for marker-assisted selection. In summary, the LDs calculated in this study for the groups of inbred rice, O. sativa and Oryza spp. accessions are most likely sufficient for marker-trait association analyses to be applied successfully to simply inherited traits. In order to assess whether SSR marker-based association mapping will be valid in a rice-breeding program, it is imperative that there are sufficiently long stretches of LD present in the breeding material under investigation.
References
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breed 19:341–356
Akkaya MS, Bhagwat AA, Cregan PB (1992) Length polymorphisms of simple sequence repeat DNA in soybean. Genetics 132:1131–1139
Bautista NS, Solis R, Kamijima O, Ishii T (2001) RAPD, RFLP and SSLP analyses of phylogenetic relationships between cultivated and wild species of rice. Genes Genet Syst 76:71–79
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Brown AHD (1979) Enzyme polymorphism in plant populations. Theor Popul Biol 15:1–42
Buckler ES, Gaut BS, McMullen MD (2006) Molecular and functional diversity of maize. Curr Opin Plant Biol 9:172–176
Chang TT (1995) Rice. In: Smartt J, Simmonds NW (eds) Evolution of crop plants. Longman, UK, pp 147–155
Chen X, Cho YG, McCouch SR (2002) Sequence divergence of rice microsatellites in Oryza and other plant species. Mol Genet Genomics 268:331–343
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Eaves IA, Barber RA, Merriman TR (1998) Comparison of linkage disequilibrium in populations from UK and Finland. Am J Hum Gen A221
Eizenga GC, Agrama HA, Lee FN, Yan W, Jia Y (2006) Characterization of newly introduced, Blast resistant rice germplasm. Crop Sci 46:1870–1878
Eizenga GC, Agrama HA, Lee FN (2007) Identifying novel resistance genes in rice wild relatives. In: Norman RJ, Meullenet J-F, Moldenhauer KAK (eds) B.R. Wells Rice Research Studies 2006. Univ Arkansas Agric Exp Stn Res Ser (in press)
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Flint-Garcia SA, Thornsberry JM, Buckler ESD (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–357
Garris A, McCouch SR, Kresovich S (2003) Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165:759–769
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Hastings A (1990) In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant Population Genetics, Breeding and Genetic Resources. Sinauer, Sunderland, MA, p 449
Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61
Hyten DL, Choi IY, Song Q, Shoemaker RC, Nelson RL, Costa JM, Specht JE, Cregan PB (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics Online:106.06970v1
Ishii T, McCouch SR (2000) Microsatellites and microsynteny in the chloroplast genomes of Oryza and eight other Gramineae species. Theor Appl Genet 100:1257–1266
Ishii T, Xu Y, McCouch SR (2001) Nuclear- and chloroplast- microsatellite variation in A-genome species of rice. Genome 44:658–666
Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C (2000) Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 290:344–447
Khush GS (1997) Origin, dispersal, cultivation and variation of rice. Plant Mol Biol 35:25–34
Kraakman ATW, Martínez F, Mussiraliev B, van Eeuwijk FA, Niks RE (2006) Linkage disequilibrium mapping of morphological, resistance, and other agronomically relevant traits in modern spring barley cultivars. Mol Breed 17:41–58
Kraakman ATW, Niks E, van den Berg PM, Stam P, van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Kraft T, Hansen M, Nilsson N-O (2000) Linkage disequilibrium and fingerprinting in sugar beet. Theor Appl Genet 101:323–326
Kruger SA, Able JA, Chalmers KJ, Langridge P (2004) Linkage disequilibrium analysis of hexaploid wheat. In: Plant & animal genomes XII conf., 10–14 January, San Diego, CA, p 321
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048
Liu K, Muse S (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lu H, Redus MA, Coburn JR, Rutger JN, McCouch SR, Tai TH (2005) Population structure and breeding patterns of 145 U.S. rice cultivars based on SSR marker analysis. Crop Sci 45:66–76
Maccaferri M, Sanguinetti MC, Noli E, Tuberosa R (2005) Population structure and long-range linkage disequilibrium in a durum wheat elite collection. Mol Breed 15:271–289
Mather DE, Hyes PM, Chalmers KJ, Eglinton J, Matus I, Richardson K, VonZitzewitz J, Marquez-Cedillo L, Hearnden P, Pal N (2004) Use of SSR marker data to study linkage disequilibrium and population structure in Hordeum vulgare: prospects for association mapping in barley. In: International barley genetics symposium, Brno, Czech Republic, 20–26 June, pp 302–307
Maurer HP, Knaak C, Melchinger AE, Ouzunova M, Frisch M (2006) Linkage disequilibrium between SSR markers in six pools of elite lines of an European breeding program for hybrid maize. Maydica 51:269–280
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Carinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Research 9:199–207
Morrell PL, Toleno DM, Lundy KE, Clegg MT (2005) Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc Natl Acad Sci USA 102:2442–2447
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc of the National Acad Sci USA 70:3321–3323
Ni J, Colowit PM, Mackill DJ (2002) Evaluation of genetic diversity in rice subspecies using microsatellite markers. Crop Sci 42:601–607
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA, Weigel D (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30:190–193
Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomanjian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, Jakobsson M, Kim S, Morozov Y, Padhukasahasram B, Plagnol V, Rosenberg NA, Shah C, Wall JD, Wang J, Zhao K, Kalbfeisch T, Schulz V, Kreitman M, Bergelson J (2005) The pattern of polymorphism in Arabidopsis thaliana. PLoS Biology 3:1289–1299
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecology Notes 6:288–295
Rakshit S, Rakshit A, Matsumura H, Takahashi Y, Hasegawa Y, Ito A, Ishii T, Miyashita NT, Terauchi R (2007) Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor Appl Genet 114:731–743
Remington DL, Thornsberry JM, Matsuola Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler IV ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A, Close TJ, Waugh R (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA 103:18656–18661
Russell JR, Booth A, Fuller JD, Baum M, Ceccarelli S, Grando S, Powell W (2003) Patterns of polymorphism detected in the chloroplast and nuclear genomes of barley landraces sampled from Syria and Jordan. Theor Appl Genet 107:413–4213
Saitou N (1987) Neighbor-joining method. Mol Biol Evol 4:406–425
Sawyer SL, Mukherjee N, Pakstis AJ, Feuk L, Kidd JR, Brooks AJ, Kidd KK (2005) Linkage disequilibrium patterns vary substantially among populations. Europ J Hum Genet 13:677–686
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Semon M, Nielsen R, Jones MP, McCouch SR (2005) The population structure of African cultivated rice Oryza glaberrima (Steud.): evidence for elevated levels of LD caused by admixture with O. sativa and ecological adaptation. Genetics 169:1639–1647
Skøt L, Humphreys MO, Armstead I, Heywood S, Skøt KP, Sanderson R, Thomas ID, Chorlton KH, Hamilton NRS (2005) An association mapping approach to identify flowering time genes in natural populations of Lolium perenne (L.). Mol Breed 15:233–245
Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet 111:723–730
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Tenesa A, Knott SA, Ward D, Smith D, Williams JL, Visscher PM (2003) Estimation of linkage disequilibrium in a sample of the United Kingdom dairy cattle population using unphased genotypes. J Anim Sci 81:617–623
Thomson MJ, Septiningsih EM, Suwardjo F, Santoso TJ, Silitonga TS, McCouch SR (2007) Genetic diversity analysis of traditional and improved Indonesian rice (Oryza sativa L.) germplasm using microsatellite markers. Theor Appl Genet 114:731–743
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler IV ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Weir BS (1996) Genetic data analysis II: materials for discrete population genetic data. Sinauer Associates, Sunderland, MA
Wright S (1978) Evolution and genetics of populations, Vol. IV. The University of Chicago Press, Chicago
Xu Y, Beachell H, McCouch SR (2005) A marker-based approach to broadening the genetic base of rice in the USA. Crop Sci 44:1947–1959
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotech 17:155–160
Acknowledgments
The support of H.A. Agrama and H.R. Refeld from the Arkansas Rice Research and Promotion Board is acknowledged. Contributions of the DB NRRC Genomics Core Facility under the direction of M.H. Jia and technical assistance of Q.P. Ho and H.R. Refeld also are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Agrama, H.A., Eizenga, G.C. Molecular diversity and genome-wide linkage disequilibrium patterns in a worldwide collection of Oryza sativa and its wild relatives. Euphytica 160, 339–355 (2008). https://doi.org/10.1007/s10681-007-9535-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-007-9535-y