
Abstract
Polymorphism over ∼26 kb of DNA sequence spanning 22 loci and one region distributed on chromosomes 1, 2, 3 and 4 was studied in 30 accessions of cultivated rice, Oryza sativa, and its wild relatives. Phylogenetic analysis using all the DNA sequences suggested that O. sativa ssp. indica and ssp. japonica were independently domesticated from a wild species O. rufipogon. O. sativa ssp. indica contained substantial genetic diversity (π = 0.0024), whereas ssp. japonica exhibited extremely low nucleotide diversity (π = 0.0001) suggesting the origin of the latter from a small number of founders. O. sativa ssp. japonica contained a larger number of derived and fixed non-synonymous substitutions as compared to ssp. indica. Nucleotide diversity and genealogical history substantially varied across the 22 loci. A locus, RLD15 on chromosome 2, showed a distinct genealogy with ssp. japonica sequences distantly separated from those of O. rufipogon and O. sativa ssp. indica. Linkage disequilibrium (LD) was analyzed in two different regions. LD in O. rufipogon decays within 5 kb, whereas it extends to ∼50 kb in O. sativa ssp. indica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice is one of the most important crops supporting more than one-third of the world population (Khush 1997). Asian cultivated rice Oryza sativa can be classified into two subspecies, ssp. indica and ssp. japonica (hereafter abbreviated as indica and japonica, respectively) based on their gross morphology and growth habitats (Oka 1988). The wild species O. rufipogon is widely accepted as the ancestor of O. sativa (Oka and Chang 1959). O. sativa is a self-pollinating species, whereas a high frequency of outcrossing is reported in O. rufipogon (Chang 1995). Africa has its own cultivated rice, O. glaberrima, which is believed to have been domesticated from the wild species O. barthii, independently from Asian Oryza. Draft sequences of the whole genomes are available for both indica (cv. 93-11; Yu et al. 2002) and japonica (cv. Nipponbare; Goff et al. 2002). More recently, a high quality whole genome sequence was published by the International Rice Genome Sequencing Project (2005). Based on the draft genome sequences, Feltus et al. (2004) compared indica and japonica sequences to study their divergence over the entire genome. According to them, 9.2% of the genome is deficient and 1.5% of the genome is rich in polymorphism between indica and japonica. Ma and Bennetzen (2004) compared a total of 37 kb of sequence of the genomes of indica, japonica and an African cultivated species, O. glaberrima, and reported that the average rate of nucleotide substitution in indica is larger than in japonica, when O. glaberrima is used as an outgroup. Nasu et al. (2002) studied DNA polymorphism in 417 regions of Oryza genome to develop SNP markers for breeding purposes. This study used three japonica, two indica and one O. rufipogon accessions as DNA materials, and showed that the O. rufipogon accession was more closely related to japonica than to indica. Nonetheless, Nasu et al. (2002) did not analyze the data from the viewpoint of population genetics. Several reports are available in which DNA polymorphism in individual loci of Oryza was studied in detail: Adh1 (Yoshida et al. 2004), Adh1 and other three loci (Zhu and Ge 2005). However, no report is available to date that studied nucleotide sequence polymorphism of Oryza at a genome-wide scale using DNA material from representative accessions of O. sativa and O. rufipogon with the aim to obtain the evolutionary history of these taxa and their DNA sequences. Such multi-locus genomics studies carried out in Drosophila melanogaster (Glinka et al 2003; Orengo and Aguade 2004) and Arabidopsis (Nordborg et al. 2005) have already provided novel insights into the evolution and population genetics of the organisms.
In order to elucidate genetic diversity and relationships of O. sativa ssp. indica, ssp. japonica and O. rufipogon, we studied DNA sequence variation in 22 loci and one region spanning over 26 kb regions of chromosomes 1, 2, 3 and 4 using 30 accessions of Oryza. This large-scale DNA polymorphism survey data was used to (1) infer the genetic relationships between O. sativa and O. rufipogon, and to (2) estimate the levels of genome-wide nucleotide diversity of O. sativa and O. rufipogon. Furthermore, the same data set was employed to (3) study the “divergence population genetics” of O. sativa ssp. indica and ssp. japonica. Finally, to evaluate the possibility of association mapping in Oryza, the extent of linkage disequilibrium (LD) in Oryza was studied for O. sativa and O. rufipogon by analyzing SNPs detected by DNA sequencing and EcoTILLING (Comai et al. 2004).
Materials and methods
DNA materials
Plant materials used in the DNA polymorphism study of the 22 loci included seven O. sativa ssp. indica, ten ssp. japonica, ten O. rufipogon, two O. glaberrima and one O. barthii accession (Table 1). Additionally, an accession of O. australiensis (IRRI acc. no. 101397) was used. For LD study, we included DNAs from other 7 O. sativa accessions and 14 O. rufipogon accessions together with the above-mentioned accessions (Suppl. Table 1). Seeds of these accessions were obtained from Kobe University and the National Institute of Genetics, Mishima, Japan. Genomic DNA was extracted from young leaves of each accession by a standard protocol.
Loci studied
A total of 22 loci (RLD3–RLD32) distributed throughout chromosomes 1, 2, 3 and 4 were selected for DNA sequence analysis (Fig. 1; Suppl. Table 2). First, EST sequences were chosen, and their corresponding genomic regions were used for DNA sequencing, so that all the studied loci were located inside or close to expressed genes. Sizes of studied loci varied from 621 bp (RLD9) to 1,201 bp (RLD25) with a mean of 897 bp. Sizes of exons contained in the loci varied from 0 bp (RLD21) to 934 bp (RLD29).

Distribution of 22 loci and two regions studied on four chromosomes of Oryza
The extent of LD was studied for two genomic regions (Regions 1 and 2) using DNAs from 24 O. sativa and 24 O. rufipogon accessions (Fig. 1; Suppl. Table 1). Region 1, studied by DNA sequencing, spans over ∼200 kb in the long arm of chromosome 3 (26.9 Mb), while Region 2, studied by EcoTILLING, covers ∼250 kb region in the long arm of chromosome 1 (34.3 Mb). These two regions were selected randomly from chromosomes 1–4. In both cases, PCR primers were designed to amplify around 1 kb regions each separated by 10–20 kb distance inside the two regions. The total size of Region 1 used for DNA sequencing was 6.2 kb, whereas that of Region 2 for EcoTILLING was 13.5 kb.
DNA sequence analysis
DNA fragments of each locus were amplified by PCR and sequenced on both strands by direct sequencing method using sequencing primers located at 300 bp interval using RISA384 (Shimadzu) and ABI377 (ABI) DNA autosequencers. Thus, most of the investigated regions were sequenced four times. Sequence readouts were assembled by Sequencher (GeneCodes) for the 22 RLD loci and AutoAssembler (ABI) for the Region 1. Quality of base-calling was checked by visual inspection of the chromatograms on Sequencher (GeneCode) and AutoAssembler (ABI). In case of discrepancy between the sequence reads of the two strands or ambiguity in base-calling, additional sequencing primers were employed to confirm the sequence. A heterozygous polymorphic site was observed only in RLD3 for the accession nos. 10-R and 11-R. For the rest of positions, we did not detect heterozygosity, presumably because most of accessions used for the study have been maintained by selfing for several generations. In the heterozygous site of RLD3 for 10-R and 11-R, the rarer nucleotide in the population was selected to represent their state for further analysis. DNA sequences were deposited in DDBJ, EMBL and GenBank DNA databases under the accession numbers, AB260142–AB260845. Sequences were aligned by CLUSTAL-W software at DDBJ, and used for further analysis. Aligned DNA sequences were analyzed by DnaSP (Rozas and Rozas 1999) and MEGA (Kumar et al. 2004) programs for estimation of population genetics parameters including π (Nei 1987) and Θ (Watterson 1975), Tajima’s D statistic (Tajima 1989) and genetic distance, D ij , between the populations i and j (Nei 1987). Hudson, Kreitman and Aguade (HKA)’s test (1987) was carried out using the program “HKA” publicly distributed by Jody Hey at Rutgers University, USA (http://www.lifesci.rutgers.edu/∼heylab). McDonald and Kreitman (MK)’s test (1991) was performed using DnaSP (Rozas and Rozas 1999). For tree construction, PHYLIP program (Felsenstein 2005) was used. A “phylogenetic” tree with the entire sequence of 22 loci was made by the neighbor-joining (NJ) method (Saitou and Nei 1987) based on pair-wise distances among the DNA sequences calculated by F84 model (Kishino and Hasegawa 1989; Felsenstein and Churchill 1996). Trees for individual loci were made by the parsimony method (Eck and Dayhoff 1966). For both NJ and parsimony trees, bootstrap resampling of sites (500 and 100 times, respectively) were applied to obtain the confidence levels of tree branches. LD measurement (r 2; Hill and Robertson 1968) and Chi-square test of statistical significance of LD for the polymorphisms detected in Regions 1 and 2 were implemented using DnaSP program (Rozas and Rozas 1999). Decay of LD with physical distance was estimated using non-linear regression according to Hill and Weir (1988) as described by Remington et al. (2001) and Ingvarsson (2005). Inter-chromosomal LD measurement, counting the number of fixed and shared polymorphism between two taxa, and Tajima’s relative rate test were carried out by using Perl scripts written by the authors.
EcoTILLING
For conducting EcoTILLING (Comai et al. 2004), a sequence of 5′-GCTACGGACTGACCTCGGAC-3′ was incorporated at the 5′-end of the locus specific upper primer and a sequence of 5′-CTGACGTGATGCTCCTGACG-3′ was attached to the 5′-end of locus specific lower primer and are referred to as UniU and UniL, respectively. UniU and UniL were also labeled with IRD-700 and IRD-800, respectively. The TILLING PCR was divided into two steps. The first PCR was carried out in a reaction volume of 20 μl having 2.25 ng genomic DNA, 4 mM each dNTPs, 0.4 U TaKaRa ExTaq and 6 μM each of tailed amplification primers. The following thermal cycling profile was used: 95°C for 2 min/35 cycles of 95°C for 1 min, 55°C for 1 min, 72°C for 1 min 30 s/72°C for 7 min. Amplification was checked on 1% agarose gel using 5 μl reaction mixture. The remaining reaction mix was diluted with 90 μl of sterile water and purified using MILLIPORE MultiScreen™ Plates as per manufacturer’s instructions. Purified amplified products were eluted by dissolving in 30 μl sterile water and further diluted 20 times (final dilution ∼1/40 × ). The second PCR was carried out in 10 μl reaction volume consisting of 2 μl diluted amplified product, 2 mM each dNTPs, 0.2 U TaKaRa ExTaq and 0.2 μM each of upper and lower primer mix. Upper primer mix was prepared by mixing labeled and unlabeled UniU primers in 3:7 ratio, and lower primer mix was made by mixing labeled and unlabeled UniL in a ratio of 2:3. The following cycling profile was used: 95°C for 2 min/35 cycles of 95°C for 1 min, 55°C for 1 min, 72°C for 1 min 30 s/72°C for 7 min/99°C for 10 min/90°C for 20 s with touch down of 0.3°C/cycle for 70 cycles. CEL-I digestion, purification and other steps were followed as described elsewhere (Colbert et al. 2001; Comai et al. 2004) except that Surveyor™ nuclease was used at 0.05 μl/reaction.
Results
Relationships between O. sativa and O. rufipogon
The nuclear genome undergoes recombination, so that genealogical relationship of DNA sequences in a locus does not necessarily correspond to those of other loci in individuals that exchange DNA by crossing and recombination. However, for taxa that are substantially differentiated from each other, we should be able to infer their relationships by “phylogenetic” analysis of DNA sequences from a large number of nuclear loci. Thus, based on DNA sequence alignment of ∼20 kb genomic sequence derived from 22 loci, a tree was constructed by the NJ method (Saitou and Nei 1987; Fig. 2). Bootstrapping resampling was applied to obtain confidence levels of clusters in the tree. The two African species O. glaberrima and O. barthii (hereafter designated as African) were well separated from the Asian species O. rufipogon and O. sativa. Separation of these African and Asian Oryza was supported 500 times in 500 bootstrapping resamplings. The japonica accessions used in the present study represented a genetically uniform group. Furthermore, japonica had two accessions of O. rufipogon (code nos. 11-R and 12-R) as sister groups (supported 421 and 425 times in 500 bootstrap resamplings, respectively), and three indica accessions (25-I, 26-I and 27-I) had one O. rufipogon (13-R) as a sister group (supported 414 times in 500 bootstrap resamplings). These results suggest that indica and japonica were independently domesticated from O. rufipogon. Genetic diversity of O. sativa was included in that of O. rufipogon. An accession (29-I) phenotypically classified as indica belonged to the cluster of japonica accessions.

A “phylogenetic” tree of Oryza species based on the sequences of 22 loci. The tree was made by NJ method (adapted from Saitou and Nei 1987) using a distance matrix among the 30 Oryza sequences. Values on branches indicate the number of times these branches were supported in 500 bootstrapping resamplings
DNA polymorphism in O. sativa and O. rufipogon
Nucleotide diversity (π) in the 22 loci (RLD3–RLD32) and one region (Region 1) spanning ∼26 kb, including both coding and non-coding regions, was 0.003 for Asian Oryza (O. sativa and O. rufipogon) (Table 2). It is noteworthy that the japonica materials studied here contained extremely low diversity (π = 0.0001). In contrast, indica had one order much larger diversity (π = 0.0027). The wild species O. rufipogon (π = 0.0034) contained a larger diversity than O. sativa (π = 0.0024). For the non-coding regions, the corresponding values were 0.0035 for Asian Oryza, 0.0028 for O. sativa, 0.0032 for indica, 0.0001 for japonica and 0.0039 for O. rufipogon. To examine whether the pattern of polymorphism observed over the whole region was in agreement with the neutral mutation hypothesis (Kimura 1983), Tajima’s test (Tajima 1989) was applied. There was a significant deviation from neutrality only in the non-coding regions of japonica (D = −1.9, P < 0.05).
Next, DNA polymorphism was studied within each locus to examine their evolutionary history. For this purpose, all the accessions of O. sativa and O. rufipogon were used together to represent the Asian population. In most cases, this treatment can be justified as the latter is considered to be a wild progenitor of the former, and these two taxa are indeed phylogenetically interrelated (Fig. 2). Nucleotide diversity of silent sites (non-coding regions and synonymous sites of coding region) for O. sativa and O. rufipogon varied largely across the loci (π = 0–0.013, Suppl. Table 3) with an average value of π = 0.003. Tajima’s test (Tajima 1989) applied to the pooled samples of O. sativa and O. rufipogon detected a statistically significant deviation from the neutral mutation expectation in RLD15 (D = 2.74, P < 0.01), RLD21 (D = −1.9, P < 0.05) and RLD27 (D = −1.83, P < 0.05). About half (11) of the loci showed positive D values, and half (10) showed negative D values.
Neutral mutation hypothesis (Kimura 1983) predicts that interspecific divergence and intraspecific polymorphism are positively correlated. HKA test (1987) uses this prediction to test whether the levels of interspecific divergence and intraspecific polymorphism are consistent to each other for more than two regions of the genome. As African Oryza species suitably serve as the outgroup for the Asian Oryza species studied here (Fig. 2), we applied the HKA test to the divergence between African and Asian species, and the polymorphism within the Asian species for multiple loci. We excluded RLD9, RLD15 and RLD22 from the test as in these loci validity of the use of African as outgroup was ambiguous (see below), so that the total number of loci used for the test was 19. Overall, HKA test did not reject the neutrality of the loci [χ2 (df = 18) = 15.4, P = 0.64]. Large χ2 values were contributed from RLD25 and RLD24, both of which had disproportionally large African–Asian divergence as compared to the polymorphism inside Asian group (Suppl. Table 3; Suppl. Fig. 1).
We tried to apply the MK’s test of neutrality to the data. However, for most of loci, this test was not applicable as the number of fixed mutations between African and Asian Oryza were too small. Even in the loci where the test was applied, we could not detect significant deviation from the neutral expectation (Suppl. Table 4).
Both the numbers of synonymous and non-synonymous segregating sites are small in japonica, followed by indica and O. rufipogon across the loci studied (Suppl. Table 5). There is no remarkable tendency that either of indica, japonica or O. rufipogon exhibits a non-synonymous/synonymous ratio different from those of other species.
It is reported that changes in the recombination frequency per unit of physical distance affect nucleotide diversity (Hudson and Kaplan 1988). To see whether there is such a tendency in Oryza, we first estimated the linkage distance (in cM) of each locus from chromosome ends by consulting a genetic map based on a cross between japonica and indica (Suppl. Table 2; Harushima et al. 1998). For each locus, we obtained linkage distances and physical distances to the two adjacent loci. Linkage distance was divided by physical distance, and the ratios from the two flanking regions were averaged to represent an estimate of recombination per site of the locus. However, there was no significant correlation observed between the levels of nucleotide diversity and the estimated recombination rates around the loci in Asian Oryza studied here (data not shown).
Divergence of indica and japonica
The two major subspecies of O. sativa, indica and japonica are thought to have diverged more than 1 million years ago (Bennetzen 2000). It is interesting to examine whether the divergence patterns of DNAs between indica and japonica are similar across the loci or not. Therefore, we calculated the distances between indica and japonica for each locus (indicated as D ij , Suppl. Table 3). They were variable (D ij = 0–0.0148) and the mean value was 0.0033, which is higher than the value estimated for the distance between the japonica cultivar Nipponbare and the indica cultivar 93-11 by whole genome sequence comparison (D ij = 0.001; Feltus et al. 2004).
In order to judge whether the rates of divergence between indica and African and that between japonica and African were different, we applied the relative rate test of Tajima (1993) for each of the 22 loci (Table 3). For the majority of loci, indica–African and japonica–African distances were not different. However, the three loci RLD9, RLD15 and RLD22 showed significant departure from equality of substitutions in a substantial number of indica and japonica accession pairs. This inequality of African–indica and African–japonica distances can be graphically seen in Fig. 3. In RLD15, African–japonica distance (D = 0.012) was almost three times larger than African–indica distance (D = 0.004), whereas in RLD9 the relationship was reverse. To understand the causes of inequality of African–japonica and African–indica distances for RLD15, RLD9 and RLD22, we constructed parsimonious trees for these loci using African species as outgroup and compared them with trees of 19 other loci (Fig. 4). Most of the 19 loci had long internal branches connecting Africa and Asian species (e.g. RLD25). However, in RLD15, the external branch connecting japonica group and other Asian accessions was exceptionally long. Similarly, in RLD9 and RLD22, the branches connecting a indica–O. rufipogon group and other Asian accessions were exceptionally long (data not shown). Tree shape like that observed for RLD15 may be obtained in two circumstances. The first is that evolutionary rates between indica and japonica lineages are not equal. The second is that the choice of outgroup for tree construction was not appropriate. To test the latter possibility, we selected O. australiensis as outgroup. O. australiensis with the EE type genome is a wild species known to be distantly related to all the Oryza species used in the present study that have the AA type genomes (Ge et al. 1999; Vaughan et al. 2003). The RLD15 region could be amplified from O. australiensis DNA by PCR, whereas the RLD9 and RLD22 regions could not. The most parsimonious tree of RLD15 incorporating O. australiensis sequence is given in Fig. 5a based on the polymorphism data in Suppl. Table 6. In this case, no long external branch specific to japonica was observed anymore. The O. australiensis–indica distance (72 mutations, D = 0.078) is equal to the O. australiensis–japonica distance (74 mutations, D = 0.081), so that we do not need to assume different evolutionary rates for indica and japonica lineages. However, quite surprisingly, the tree topology obtained with O. australiensis as outgroup indicated that japonica sequences are separated from African sequences and the rest of Asian sequences (461 times out of 500 bootstrap replications), which was in striking contrast to the tree obtained from the 22 loci (Fig. 2). It is noteworthy that an O. rufipogon accession 11-R clusters with japonica in RLD15. This result suggests that RLD15 region of japonica has a history different from other genomic regions.

Relationship between African Oryza–O. sativa ssp. japonica distance (D FJ—y-axis) and African Oryza–O. sativa ssp. indica distance (D FI—x-axis)

Most parsimonious trees for the two loci RLD25 and RLD15 constructed by using sequences of African Oryza (O. glaberrima—2-G, 3-G and O. barthii—1-B) as outgroup. The code numbers correspond to those given in Table 1 The numbers on branches are bootstrapping probabilities (%)

Evolutionary history of RLD15 locus. a The most parsimonious tree of RLD15 using O. australiensis as outgroup. The values on branches indicate the number of times the branches were supported after 500 bootstrap resamplings. b and c Organismal tree (black line) and DNA tree (red line) for the “trans-species polymorphism” hypothesis (b) and the “introgression hypothesis” (c)
DNA polymorphism in Asian Oryza can be classified into those fixed in one of the groups, indica, japonica and O. rufipogon, or shared between two of these groups. There were 69 shared and zero fixed mutations detected between indica and O. rufipogon, whereas only three shared and six fixed mutations between japonica and O. rufipogon (Suppl. Table 7). The number of fixed mutations in japonica is disproportionally larger than that of indica.
Coding site divergence between indica and japonica
Next, we focused on divergence on coding sites of each locus. For each locus, the most parsimonious tree was constructed, and for the non-synonymous substitutions, the ancestral and derived states were inferred for indica, japonica and O. rufipogon (Table 4). We excluded the three loci RLD9, RLD15 and RLD22 from the analysis as the use of African as the outgroup in these loci are not secure (see above). There was only one amino acid change that was almost fixed in indica (RLD31-430; Ser → Leu). In contrast, there were six amino acid changes that were fixed or almost fixed in japonica. Synonymous mutations can be divided into “preferred” and “unpreferred” mutations (Akashi 1995). We can classify the derived mutations to “preferred” or “unpreferred” by consulting a table of preferred codons in Oryza (Kawabe and Miyashita 2003). Both indica and japonica had one fixed substitution in “preferred” to “unpreferred” direction and one in “unpreferred to preferred” direction, respectively, and no difference in the tendency of synonymous mutations detected between indica and japonica.
Extent of linkage disequilibrium in Oryza
Linkage disequilibrium is a measure of non-random association of alleles of different loci in a population. The degree of LD is determined by the recombination rate between the loci, which is governed by the mating system (inbreeding vs. outcrossing) of the organisms, by epistatic selection of two distant loci, and by migration (Gillespie 2004). Knowledge on the levels of LD is practically important to evaluate the feasibility of association mapping to identify the genomic regions containing loci controlling agronomic performance of the crops (Gupta et al. 2005).
Significant LD can arise simply by the mixture of genetically distant groups in the population. To evaluate this possibility, we measured LD among all the loci distributing over the four chromosomes. The rational behind is that LD detected for two loci from different chromosomes are not caused by genetic linkage but by population substructuring. As is evident from Suppl. Table 8a, there was almost no LD between two loci from different chromosomes in O. rufipogon, whereas there was extensive LD in O. sativa (Suppl. Table 8b). This LD of O. sativa across the genome is caused by the extensive divergence between indica and japonica, as the significant LD substantially decreased if indica and japonica were studied separately (Suppl. Table 8c, d). The level of DNA polymorphism in japonica was too low to derive a meaningful picture on LD in this subspecies. Therefore, we focused on intra-chromosomal LD only for O. rufipogon and O. sativa ssp. indica for the rest of study.
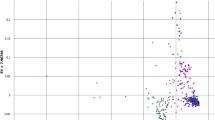
To study the LD levels in Oryza, we analyzed two genomic regions with two different methods of SNP detection: DNA sequencing and EcoTILLING, a novel high-throughput technique for detection of DNA polymorphism (Comai et al. 2004). The results are presented separately for O. rufipogon and indica in Fig. 6. In O. rufipogon, DNA sequencing of Region 1 detected 95 sites useful for LD measurement (Fig. 6a). The majority (83%) of site-pairs had the r 2 values <0.2. Statistically significant LD was detected in 875 pairs among 4,552 comparisons (19%). Similarly, EcoTILLING of Region 2 detected 94 informative SNPs for LD measurement, and r 2 values were <0.2 in 84% of the cases (Fig. 6b). Significant LD was observed in 892 pairs among 4,406 comparisons (20%). In O. rufipogon, the levels of LD in Regions 1 and 2 were similar. As the regression curve indicates, LD rapidly decays in O. rufipogon. Examination of intra-locus LD for 22 loci (Suppl. Table 8a) shows that only the three loci RLD15, RLD24 and RLD32 have average r 2 values more than 0.5, and the rest exhibited low values of r 2. These results suggest that LD in O. rufipogon decays within <5 kb.

Extent of LD in Oryza. Each spot represents distance (bp) between the two polymorphic sites (x-axis) and LD of them as measured by r 2 (y-axis). Regression curves were fitted according to Hill and Weir (1988). Each graph corresponds to DNA polymorphism detected in a O. rufipogon Region 1 as studied by DNA sequencing; b O. rufipogon Region 2 as studied by EcoTILLING; c O. sativa ssp. indica Region 1 as studied by DNA sequencing and d O. sativa ssp. indica Region 2 as studied by EcoTILLING
For O. sativa ssp. indica, DNA sequencing of Region 1 identified 62 SNPs useful for LD measurement (Fig. 6c), and 789 out of 1,893 comparisons (42%) were statistically significant (P < 0.05). EcoTILLING of Region 2 identified 67 SNPs (Fig. 6d), and 738 out of 2,211 comparisons (33%) were significant. Levels of LD were similar in Region 1 and Region 2 (Fig. 6c, d). LD decay of indica is slow as compared to O. rufipogon, and the regression curve indicates that substantial LD extends over 50 kb region in this subspecies.
Discussion
Relationship between O. sativa ssp. indica, ssp. japonica and O. rufipogon
Genetic relationships between O. sativa and O. rufipogon were studied by constructing a tree using the entire 20 kb sequence of 22 loci (Fig. 2). The tree indicated that O. sativa accessions do not form a unique group separated from O. rufipogon accessions. Rather, most O. sativa accessions have O. rufipogon accessions as sister groups. This finding supports the hypothesis that O. sativa cultivars were independently domesticated from wild O. rufipogon in multiple occasions (Second 1982; Ishii et al. 1988; Cheng et al. 2003; Bautista et al. 2001). Over the 22 loci and Region 1 in the genome, nucleotide diversity of non-coding regions in O. sativa and O. rufipogon is low (π = 0.003; Table 2). This is half of the value obtained for the genome of Arabidopsis (π = 0.006, Nordborg et al. 2005). It is noteworthy that nucleotide diversity in japonica was extremely low (π = 0.0001; Table 2). This result indicates that our japonica samples studied here have a short coalescence time, and they must originate from a small number of founders that presumably were genetically close to O. rufipogon genetically close to the 11-R accession used in the present study.
Divergence population genetics of O. sativa ssp. indica and ssp. japonica
The present study confirmed that both O. sativa ssp. indica and ssp. japonica were derived from O. rufipogon. It was also shown that indica contains more than an order of magnitude larger genetic diversity as compared to japonica. In other words, the effective size of indica population is much larger than that of japonica. This large difference in population effective size prompted us to compare the nature of mutations in indica and japonica with reference to O. rufipogon. Such approach was named “divergence population genetics” by Kliman et al. (2000) and has been applied to Drosophila species, where it provided fruitful results. When we compared the shared and fixed polymorphism between the pairs of indica, japonica and O. rufipogon, there was a remarkable difference between indica–O. rufipogon and japonica–O. rufipogon pairs (Suppl. Table 7), namely all the polymorphisms in indica–O. rufipogon comparison was shared between the two taxa, whereas there were only three shared polymorphism and six fixed mutations between japonica and O. rufipogon. This result suggests that japonica has accumulated unique mutations after its divergence from O. rufipogon. Furthermore, patterns of derived non-synonymous mutations are also different between japonica and indica (Table 4). The japonica samples contained a larger number of non-synonymous fixed mutations as compared to indica. This finding can be explained by two possibilities: (1) positive selection, presumably artificial selection, imposed on the japonica genome, (2) accumulation of slightly deleterious mutations in the japonica genome resulting from its small population size. As it is highly improbable that we detected trace of positive selection in randomly selected tiny portion of the genome as studied here, we prefer the possibility (2). It is theoretically established that slightly deleterious mutations have a larger probability of fixation in a population with a smaller effective size than that with a larger effective size (Ohta 1976). Higher allozyme heterozygosity in contrast to low nucleotide diversity has been reported in D. melanogaster in comparison to D. simulans (Aquadro et al. 1988). The authors suggested that this may be explained by a smaller effective size of D. melanogaster than that of D. simulans. A study of divergence and polymorphism of three Drosophila species, D. simulans, D. mauritiana and D. sechellia (Kliman et al. 2000) showed that D. sechellia with the lowest effective population size had accumulated larger number or mutations than other species, and that at synonymous sites, D. sechellia accumulated significant excess of unpreferred substitutions. Kliman et al. (2000) suggested that this pattern of divergence in D. sechellia is explained by the accumulation of slightly deleterious mutations in D. sechellia with the small effective size. We hypothesize that a similar mechanism may explain the larger number of fixed non-synonymous polymorphism in japonica as compared to indica.
Heterogeneity in DNA polymorphism and genealogy across the loci
Nucleotide diversity of 22 loci of Asian Oryza (pooled samples of O. sativa and O. rufipogon) is heterogenous (Suppl. Table 3). Tajima’s D also varied across the loci. RLD15 showed statistically significant positive value of Tajima’s D, which is caused by the large divergence between japonica and indica sequences in this locus (see below). In contrast, RLD21 and RLD27 had significant negative values of Tajima’s D, which is caused by a larger number of singleton mutations in these loci (7/10 for RLD21 and 6/9 for RLD27). Genealogy of DNA also varied across the loci (Fig. 4). These results suggest that different loci have been placed under different evolutionary forces including natural selection. In the present study, we could not detect significant relationship between the recombination rate and the amount of DNA polymorphism as observed in D. melanogaster by Begun and Aquadro (1992). The current study was carried out over very large physical distances, with a rough estimation of recombination per kilobase, so that such a correlation may be addressed more appropriately by a better estimation of recombination rate (see Glinka et al. 2003; Orengo and Aguade 2004). O. sativa ssp. indica and ssp. japonica are well-separated entities of rice cultivars, so that it is meaningful to compare the divergence pattern between these two subspecies. For most of the loci studied, the divergence between indica and African is similar to that between japonica and African (Fig. 3), and we could not detect a general tendency that the indica–African divergence is larger than japonica–African divergence as noted by Ma and Bennetzen (2004).
Genealogy of RLD15 locus
Genealogical analysis of RLD15 locus of O. sativa and O. rufipogon using African species as outgroup generated a tree with a long branch connecting japonica and other Asian Oryza (Fig. 4). When O. australiensis, a more distantly related species, was used as outgroup, this long branch disappeared, so that we do not need to assume an accelerated evolution in the japonica lineage. However, in this case, japonica formed a group outside other Asian species that clustered with African species (Fig. 5a). The discrepancy of tree topologies in Figs. 2 and 5a can be explained by either of the two hypotheses (Fig. 5b, c).
The first one, “trans-species polymorphism hypothesis,” postulates that the bifurcation of RLD15 DNA lineages occurred in a population of the common ancestor of African species as well as Asian O. rufipogon (Fig. 5b). The two DNA lineages (alleles) were maintained in O. rufipogon population for a substantial time before each of them was inherited by O. sativa indica and japonica, respectively. Such DNA polymorphisms could be maintained in a population for a long time by (1) strong balancing selection imposed on the tightly linked genomic region, or by (2) mere chance. An extreme case of balancing selection is diversifying selection as exemplified in MHC genes in primates (Lawlor et al. 1988) and self-incompatibility genes in plants (Ioerger et al. 1991). Such “trans-species polymorphism” occurs by chance as well when the divergence of the species is quite recent as seen in Drosophila (Hey and Kliman 1993).
The second hypothesis is the “introgression hypothesis.” In this case, the RLD15 region of japonica species was replaced by recombination with a DNA sequence from a yet unknown species (species X) that has diverged from the AA genome group of O. sativa, O. rufipogon, O. glaberrima and O. barthii. If this is the case, the introgression should have occurred before the domestication of japonica as an O. rufipogon accession (11-R) is also included in this japonica cluster.
At the moment we have no good reason to choose one from these two hypotheses. These two mechanisms may together simultaneously function to result in the observed patterns. An extended study of DNA polymorphism around RLD15 using materials including Oryza taxa with the genomes B, C and D (Vaughan et al. 2003) will clarify the evolutionary history of this genomic region of japonica. Feltus et al. (2004) compared genome sequences of indica and japonica rice, and noted that there are highly divergent as well as very similar regions between the two sequences. Interestingly, the chromosome region harboring RLD15 corresponds to the region exhibiting a high genetic divergence between indica and japonica DNA sequences (Feltus et al. 2004). This suggests a possibility that the rest of genomic regions showing large divergence between indica and japonica sequences could have a similar history as RLD15. The “phylogenetic” analysis using the DNA sequences of 22 loci included RLD15. Removal of this locus from the analysis did not change the tree topology as shown in Fig. 2 (data not shown) as its contribution among the entire sequence was small.
Linkage disequilibrium in Oryza
DNA sequencing and EcoTILLING in the two genomic regions of Oryza showed that levels of LD are remarkably different between O. rufipogon and O. sativa ssp. indica (Fig. 6). LD in O. rufipogon decays within 5 kb, whereas it extends to 50 kb in O. sativa ssp. indica. This difference in the extent of LD may partly be explained by the breeding behavior of the species. Lower levels of LD in O. rufipogon are expected because O. rufipogon experiences higher rate of outcrossing as opposed to O. sativa which is strictly a self-pollinated crop (Khush 1997). In maize, a cross-pollinated crop, LD declines fast within a distance of 1,500 bp (Remington et al. 2001), whereas in Arabidopsis, a selfing species, LD extends to approximately 20 kb (Nordborg et al. 2002, 2005). For practical purposes, the low LD in O. rufipogon would be advantageous for association-based cloning of genes controlling phenotypic traits by using a large number of markers. In O. sativa ssp. indica, we will be able to use markers spaced 50 kb apart on average to carry out a rough agronomic trait mapping and marker-assisted selection.
In conclusion, this large-scale DNA polymorphism study of O. sativa and its wild relatives revealed the origin and divergence of cultivated rice in unprecedented details. In view of rapid development of high-throughput sequencing and SNP-finding technologies, in the near future we will be able to address the origins of indica and japonica rice in further detail, and identify the causal mutations that allowed their domestication from wild progenitors.
References
Akashi H (1995) Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics 139:1067–1076
Aquadro CF, Lado KM, Noon WA (1988) The rosy region of Drosophila melanogaster and Drosophila simulans. I. Contrasting levels of naturally occurring DNA restriction map variation and divergence. Genetics 119:875–888
Bautista NS, Solis R, Kamijima O, Ishii T (2001) RAPD, RFLP and SSLP analyses of phylogenetic relationships between cultivated and wild species of rice. Genes Genet Syst 77:71–79
Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in Drosophila melanogaster. Nature 356:519–520
Bennetzen JL (2000) Comparative sequence analysis of plant genomes: microcolinearity and its many exceptions. Plant Cell 12:1021–1029
Chang TT (1995) Rice. In: Smartt J, Simmonds NW (eds) Evolution of crop plants. Longman, UK, pp 147–155
Cheng C, Motohashi R, Tsuchimoto S, Fukuta Y, Ohtsubo H, Ohtshubo E (2003) Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Mol Biol Evol 20:67–75
Colbert T, Till BJ, Tompa R, Rynold S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S (2001) High-throughput screening for induced point mutations. Plant Physiol 126:480–484
Comai L, Young K, Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Henikoff S (2004) Efficient discovery of DNA polymorphisms in natural populations by EcoTILLING. Plant J 37:778–786
Eck RV, Dayhoff MO (1966) In: Dayhoff MO (ed) Atlas of protein sequence and structure. National Biomedical Research Foundation, Silver Springs
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Felsenstein J, Churchill GA (1996) A Hidden Markov Model approach to variation among sites in rate of evolution. Mol Biol Evol 13:93–104
Feltus FA, Wan J, Shulze SR, Estill JC, Jiang N, Paterson AH (2004) An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res 14:1812–1819
Ge S, Sang T, Lu BR, Hong DY (1999) Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc Natl Acad Sci USA 96:14400–14405
Gillespie JH (2004) Population genetics. A concise guide, 2nd edn. The Johns Hopkins University Press, Baltimore, pp 214
Glinka S, Ometto L, Mousset S, Stephan W, De Lorenzo D (2003) Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics 165:1269–1278
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa ssp. japonica). Science 286:91–100
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A, Kajiya H, Huang N, Yamamoto K, Nagamura Y, Kurata N, Khush GS, Sasaki T (1998) A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148:479–494
Hey J, Kliman RM (1993) Population genetics and phylogenetics of DNA sequence variation at multiple loci within the Drosophila melanogaster species complex. Mol Biol Evol 10:804–822
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231
Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78
Hudson RR, Kaplan NL (1988) The coalescent process in models with selection and recombination. Genetics 111:147–164
Hudson RR, Kreitman M, Aguade M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 136:1329–1340
Ingvarsson PK (2005) Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European Aspen (Populus tremula L., Salicaceae). Genetics 169:945–953
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ioerger TR, Clark AG, Kao TH (1991) Polymorphism at the self-incompatibility locus in Solanaceae predates speciation. Proc Natl Acad Sci USA 87:9732–9735
Ishii T, Terauchi T, Tsunewaki K (1988) Restriction endonuclease analysis of chloroplast DNA from A-genome diploid species of rice. Jpn J Genet 63:523–536
Kawabe A, Miyashita NT (2003) Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet Syst 78:343–352
Khush GS (1997) Origin, dispersal, cultivation and variation of rice. Plant Mol Biol 35:25–34
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, London
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol 29:170–179
Kliman RM, Andolfatto P, Coyne JA, Depaulis F, Kreitman M, Berry AJ, McCarter J, Wakely J, Hey J (2000) The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics 156:1913–1931
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P (1988) HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature 335:268–271
Ma J, Bennetzen JL (2004) Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci USA 101:12404–12410
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Nasu S, Suzuki J, Ohta R, Hasegawa K, Yui R, Kitazawa N, Monna L, Minobe Y (2002) Search for and analysis of single nucleotide polymorphisms (SNPs) in rice (Oryza sativa, Oryza rufipogon) and establishment of SNP markers. DNA Res 9:163–171
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA, Weigel D (2002) The extent of linkage disequilibrium in Arabisopsis thaliana. Nat Genet 30:190–193
Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, Jakobsson M, Kim S, Morozov Y, Padhukasahasram B, Plagnol V, Rosenberg NA, Shah C, Wall JD, Wang J, Zhao K, Kalbfleisch T, Schulz V, Kreitman M, Bergelson J (2005) The pattern of polymorphism in Arabidopsis thaliana. PLOS Biol 3:1289–1299
Ohta T (1976) Role of very slightly deleterious mutations in molecular evolution and polymorphism. Theor Popul Biol 10:254–275
Oka HI (1988) Origin of cultivated rice. Japan Scientific Societies Press, Tokyo/Elsevier, Amsterdam
Oka HI, Chang WT (1959) The impact of cultivation on populations of wild rice, Oryza sativa f. spontanea. Phyton 13:105–117
Orengo DJ, Aguade M (2004) Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattern of variation and distance to coding regions. Genetics 167:1759–1766
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES 4th (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Rozas J, Rozas R (1999) DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15:174–175
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Second G (1982) Origin of the genic diversity of cultivated rice (Oryza spp.): study of the polymorphism scored at 40 isozyme loci. Jpn J Genet 57:25–57
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tajima F (1993) Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135:599–607
Vaughan DA, Morishima H, Kadowaki K (2003) Diversity in the Oryza genus. Curr Opin Plant Biol 6:139–146
Watterson GA (1975) On the number of segregating sites in genetic models without recombination. Theor Popul Biol 7:256–276
Yoshida R, Miyashita NT, Ishii T (2004) Nucleotide polymorphism in the Adh1 locus region of the wild rice Oryza rufipogon. Theor Appl Genet 109:1406–1416
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H (2002) A draft sequence of the rice genome (Oryza sativa L .ssp. indica). Science 296:79–92
Zhu Q, Ge S (2005) Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol 167:249–265
Acknowledgments
SR conducted this work under financial support from Japanese Society for Promotion of Sciences, Tokyo. RT thanks B. Till, L. Comai and S. Henikoff, Washington University, Seatle, USA for training him with TILLING technique. We thank N. Kurata, National Institute of Genetics, Mishima, Japan for providing rice seeds, and J. Rozas, University of Barcelona, Spain for guiding us with the DnaSP program, two anonymous reviewers and M. Morgante, University of Udine, Italy for valuable comments to the earlier version of the manuscript. This work was supported by the “Program for Promotion of Basic Research Activities for Innovative Biosciences” (Japan), “Iwate University 21st Century COE Program: Establishment of Thermo-Biosystem Research Program” and “JSPS grant no. 18310136” to RT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Morgante.
Electronic supplementary material
Below is the link to the electronic supplementary material
Fig7
Supplementary Fig. 1. Results of multilocus HKA test. Contributions to the overall χ2 test static by the polymorphism and divergence observations for each locus. Squares indicate divergence between African and Asian species, and circles correspond to polymorphism within Asian species. If the observed value was greater than the expected, then the point is placed above the line; otherwise it is placed below the line (JPG 74 kb)
122_2006_473_MOESM3_ESM.doc
Supplementary Table 3. DNA polymorphism of O. sativa and O. rufipogon for silent positions (non-coding and coding synonymous sites) at the 22 loci (DOC 34 kb)
122_2006_473_MOESM5_ESM.doc
Supplementary Table 5. Number of segregating sites within all positions (coding and non-coding positions) for Asian Oryza species (DOC 25 kb)
122_2006_473_MOESM6_ESM.doc
Supplementary Table 6. DNA variation in RLD15 locus (Only non-singleton mutations are shown. There were 67 singleton mutations specific to O. australiensis) (DOC 30 kb)
122_2006_473_MOESM7_ESM.doc
Supplementary Table 7. Shared polymorphism and fixed differences among O. sativa ssp. indica, ssp. japonica and O. rufipogon (DOC 33 kb)
Rights and permissions
About this article
Cite this article
Rakshit, S., Rakshit, A., Matsumura, H. et al. Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor Appl Genet 114, 731–743 (2007). https://doi.org/10.1007/s00122-006-0473-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0473-1