
Abstract
The genetic diversity of Egyptian rice has received diminished attention recently, resulting in a weakened genetic foundation that renders it more vulnerable to environmental fluctuations. Developing durable rice cultivars necessitates the ongoing exploration of genetic resources and the utilization of diverse germplasm. The examination of rice diversity was conducted by researchers utilizing 27 rice genotypes and 23 polymorphic microsatellite markers. The microsatellite comprises 106 alleles that have been observed (Na), with an effective allele count of 67.36 (Ne). The average number of alleles per locus is 4.61, with a standard deviation of 2.93. On average, the values of polymorphism information content (PIC) were found to be 0.580. A noteworthy correlation was observed between the number of alleles and both gene diversity (r = 0.713) and polymorphic information content (PIC) (r = 0.785). The results of our study provide empirical evidence supporting the existence of both the P1 and P2 subgroups; P1 comprises twelve distinct rice genotypes, while P2 encompasses fifteen different rice genotypes. The analysis of molecular variance determined that individual variations contributed to 34.22% of the observed variance, while population differences accounted for the remaining 65.78%. The gene flow coefficient was determined to be 0.481, whereas the fixation index exhibited a statistically significant value of 0.342. The presence of admixtures within the population suggests a significant likelihood of gene transfer between different genotypes. The principal component analysis (PCA) encompassed 27 components, with only the initial four components (PC1, PC2, PC3, and PC4) regarded as variation representatives. In that order, the initial four components exhibited the highest eigenvalues, specifically 18.413, 10.759, 7.618, and 7.031. The findings underscore the importance of investigating and establishing connections between genetic diversity and developing Egyptian rice breeding and enhancement strategies. Selecting genotypes as possible donors in a crop breeding approach to improve rice quality requires examining genetic diversity within populations. The distribution of alleles within subpopulations reveals valuable loci for genetic diversity research. This study contributes to developing environmentally sustainable cultivars by providing insights into the mechanisms of gene flow and genetic structure in Egyptian rice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice, scientifically known as Oryza sativa L., holds considerable agricultural importance as it is a primary food source for approximately 50% of the global population. Furthermore, it ranks second in crop consumption, following wheat (Babu et al. 2014). Rice, the second most widely consumed cereal, is a significant dietary staple for approximately 3.5 billion individuals globally, contributing to about 20% of their caloric intake. According to recent projections, there is an anticipated increase in global rice consumption to reach 852 million tonnes by the year 2035. This surge in demand can be attributed to the expected growth in the global population within the next quarter of a century (Khush 2013). Egypt serves as the primary source of sustenance for a population exceeding 105.82 million individuals. Egypt is positioned at the 18th spot globally regarding its ranking among the largest nations in rice production. The cultivation of rice receives greater emphasis in the lower region of the Nile River.
Nevertheless, the cultivation of rice in Egypt has been constrained as a consequence of inadequate water resources. Moreover, it is worth noting that there has been a substantial decline in the diversity of rice species over the past few centuries (Choudhary et al. 2013). Consequently, the development of novel rice cultivars assumes paramount importance in meeting the demands of the global population and mitigating the challenges posed by biotic and abiotic stresses. Implementing crop advancement strategies can result in higher crop yields and enhanced resilience against abiotic stressors (Rai et al. 2023). The success of breeding projects may depend on genetic diversity within rice genotypes.
Consequently, the pursuit of greater cultivar variability necessitates examining and quantifying genetic diversity across a wide range of genetic lineages. The existing diversity within plant populations can be harnessed to facilitate the development of cultivars that exhibit enhanced tolerance to abiotic stressors such as drought and salinity, which are prevalent abiotic factors in the Egyptian context. To effectively address genetic diversity, two distinct approaches can be employed: (1) the utilization of agronomic characteristics and (2) the assessment of DNA variations through molecular techniques.
Some limitations of assessing genetic diversity based on physiological traits include the extensive time required, the need to utilize multiple sites and years, the impact of environmental factors, and the insufficient resolving power to distinguish highly similar genotypes. In contrast, using molecular DNA markers has yielded significant progress in evaluating genetic diversity in various crop species (Mourad et al. 2019; Mohanty et al. 2021; Naaz et al. 2022; Safhi et al. 2022; Ibrahim et al. 2023a, b). Microsatellites, known as simple sequence repeats (SSRs), are highly polymorphic DNA markers widely employed in species diversification research (Abd El-Moneim 2021; Hassan and Hama-Ali 2022; Mesfer ALshamrani et al. 2022; Ibrahim et al. 2023a, b). In order to investigate the genetic variability in rice, microsatellite markers are utilized due to their co-dominant nature, abundance in the genome, informativeness, repeatability, reliability, low cost, and ability to exhibit a high rate of polymorphism (Borba et al. 2010; Hassan and Hama-Ali 2022).
In previous research, cluster analysis was employed to ascertain the interconnectedness between genotypes, aiming to characterize the biological diversity present. The progress in population structures has led to the development of diverse software applications that facilitate a more thorough comprehension of the genetic makeup of a population. One of the key components of these programs is structure, which produces notable clusters based on the presence of Hardy–Weinberg disequilibrium and linkage disequilibrium (LD) resulting from population admixture (Pritchard et al. 2000; Safhi et al. 2022). Considering linkage disequilibrium (LD) among genotypes has enhanced the quality of clustering findings (Falush et al. 2003; EL-Mansy et al. 2021; Essa et al. 2023). This approach facilitates the efficient utilization of genetic diversity in breeding endeavors to develop novel cultivars harboring genes associated with enhanced productivity and heightened resilience to biotic and abiotic stressors. This study aims to investigate the population structure and gene flow of 27 Egyptian rice genotypes using microsatellite markers, assess the level of genetic variability among these genotypes, and compare genetic characteristics between subpopulations using microsatellite markers.
Materials and methods
Plant materials
An extensive and diverse collection of 27 rice genotypes was used for the analysis. These genotypes were obtained from the Genetic Stocks Oryza Collection, the USA, and the Agricultural Research Center (ARC), Giza, Egypt. A list of the rice genotypes, accession number, source of seeds, and subspecies group is presented in Table S1.
Sample preparation and DNA extraction
Total genomic DNA was extracted from young leaves of 8-weeks-old seedlings growing under a greenhouse for five-seedling individuals for each genotype used as a genetic pool in the Faculty of Science, Arish University 2022. DNA extraction was performed according to Salem et al. (2004).
PCR amplification of microsatellite markers
Twenty-three microsatellite markers representing chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 were employed to assay genetic variation for all genotypes based on the Rice Genes Database (http://gramene.org). Detailed information on the microsatellite markers is given in Table S2. In a total volume of 25 μl, the Reaction mixture comprised 50–100 ng template DNA, 0.2 mM dNTPs (Qiagen), 1.5 mM MgCl (Qiagen), 250 nM forward primer, 250 nM reverse primer (Applied Biosystems, ThermoFisher), 2.5 μl PCR buffer (10X) and 1U Taq DNA polymerase (Qiagen). The following program was used to perform PCR: 5 min at 94 °C, followed by 35 cycles for 1 min at 94 °C, 1 min at primer’s annealing temperature accordingly, and 2 min at 72 °C, with a final extension of 5 min at 72 °C, as described by Wu and Tanksley 1993. The amplification products were resolved on 10% polyacrylamide denaturing gels (PAGE). A binary data matrix was used to score the gels. The presence (1) and absence (0) were recorded for each variation of alleles.
Genetic diversity estimation
A dendrogram of rice genotypes was generated based on the unweighted pair group method with arithmetic mean (UPGMA) method via Power Marker DARWIN version 5 was used to calculate the variation in the experimental sample using allele scoring data (Perrier and Jacquemoud-Collet 2006). A dendrogram of rice genotypes was generated based on the unweighted neighbor-joining method using the Dice similarity index method via DARWIN version 5 (Perrier and Jacquemoud-Collet 2006). The observed heterozygosity (Ho) and expected heterozygosity (He), the number of alleles per locus, the number of polymorphic loci, as well as Shannon (I) and Nei information indices (between and within the populations) were determined by using POPGENE version 1.32 (Falush et al. 2003; Yeh et al. 1997). The polymorphic information content (PIC), gene diversity (GD), and allelic frequency for all markers were analyzed by Power Marker version 3.25 (Lui 2005).
Population structure analysis
A basic STRUCTURE model (Stanford University, Stanford, California) described by Pritchard et al. (2000) was used to estimate and analyze population structure. It uses a correlated allele frequency and admixtures model (Falush et al. 2003; Hubisz et al. 2009). The values of K were set from 1 to 10, and each value with ten repeats. The length of the burn-in period was set at 20,000, and 2,00,000 Monte Carlo Markov chain runs were set to estimate the value of the K. Structure Harvester software program, which was used to determine the final population based on second-order data (Earl and Von Holdt 2012).
Analysis of molecular variance (AMOVA) and venn diagram
The ARLEQUIN version 3.114 was implemented for calculating molecular variance (AMOVA). The VENNEY 2.1 software program was used to generate the Venn diagram and to determine shared germplasm among populations and hierarchical groups (Oliveros 2007). Using R Software, Principal component analysis was used to illustrate the relationships among the studied rice genotypes.
Results
Genetic variability of microsatellite loci among rice genotypes
In order to evaluate the genetic variability in rice, a total of 23 microsatellite markers specific to rice were employed. All microsatellite markers exhibited polymorphism, and the dataset contained no duplication. A comprehensive analysis revealed the presence of 106 distinct alleles among a set of 27 rice genotypes. The alleles identified in this study exhibited a range of two for RM144, RM151, RM215, and RM277 to eight for RM11, resulting in an average of 4.61 alleles per locus. The amplified fragments exhibited a range of lengths from 78 to 264 base pairs (Table 1).
In this study, the efficacy of the microsatellite markers was evaluated by measuring parameters such as the number of observed alleles (N), number of effective alleles (Ne), polymorphic information content (PIC), Nei gene diversity, and Shannon index (I) across all genotypes. The data about the bands generated are presented in Table 1. The effective alleles (Ne) observed at each locus exhibited a range of 1.87 to 4.53, with a mean value of 2.93. Similarly, the genetic diversity (GD) observed per locus varied between 0.466 (RM151) and 0.779 (RM19). The range of He values observed in this study varied from 0.475 (RM151) to 0.794 (RM19). The Shannon (I) and Nei information indices exhibited a range of 0.659 to 1.690 and 0.466 to 0.779, respectively, with mean values of 1.193 and 0.631. In addition, the values of PIC varied from 0.358 (RM151) to 0.751 (RM19), with an average of 0.580. The microsatellite marker RM19, which possesses a polymorphic information content (PIC) value of 0.751, exhibited a higher level of polymorphic information. The marker indicates a maximum PIC value of RM19 and displays six distinct alleles across the various genotypes, as depicted in Table 1. The study revealed a significant and robust Pearson correlation coefficient (r) between the number of alleles and both polymorphic information content (PIC) (r = 0.785**, P < 0.01) and gene diversity (r = 0.713**, P < 0.01) (Figure S1 and S2).
Population structure analysis
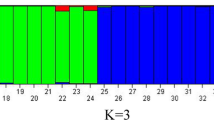
The utilization of the STRUCTURE program facilitated the assessment of the genetic structure of the individual rice samples, resulting in the most dependable differentiation of the various genotypes. The population genetic structure analysis of 27 rice genotypes was conducted using microsatellite genotyping data from the STRUCTURE program. The research offers valuable data for establishing the suitable population structure of the Egyptian rice genotypes. The genetic composition analyzed in this research was derived from a total of 106 distinct alleles that were distributed among 27 different germplasm genotypes. The optimal number of subpopulations (K) was determined using the Structure Harvester algorithm, and the results were presented in Fig. 1, indicating that the most suitable value for K was 2 (K = 2). As a result, the subpopulation results of K = 2 were selected using the STRUCTURE software (Fig. 1).

The distribution of delta K (ΔK) for different numbers of subpopulations to determine the true number of subpopulations (K). The sharp peak of DK at K = 2 suggests two subpopulations
The red cluster, denoted as P1, comprises twelve distinct rice genotypes, while the green cluster, designated as P2, encompasses fifteen distinct rice genotypes. Concerning the microsatellite diversity parameters, the levels of heterozygosity (Ho) and effective population size (Ne) did not exhibit significant diversity in P1, P2, and P1P2, as indicated in Table 2. The level of heterozygosity (He) observed in the P2 subpopulation was relatively higher when compared to the P1 and P1P2 subpopulations. Following this, when the K value was set to 3, 27 genotypes were classified into three distinct subpopulations, namely P1, P2, and P3. It is worth noting that in these subpopulations, the number of admixtures exceeded that observed when K was set to 2. Figure 2 illustrates a visual representation of subpopulations at two distinct K values. At a K value of 3, the subpopulations P1, P2, and P3 consisted of 11, 5, and 11 genotypes, respectively. At a K value of 2, except admixtures, all genotypes assigned to the subpopulations (P1 and P2) were deemed pure as their membership probability exceeded 0.90 (Fig. 2).

Population structure estimates for 27 Egyptian rice genotypes using microsatellite markers. a Two sub-populations identified with STRUCTURE analysis. b Three sub-populations identified with STRUCTURE analysis. The y-axis represents sub-group membership, whereas the x-axis represents genotype
Additionally, to analyze the diversity within subpopulations, the scoring data was aggregated in a manner specific to each subpopulation (P1, P2, and P1P2). The mean number of distinct alleles in subpopulation P2 (5.93) exhibited a slight increase compared to subpopulation P1 (5.10) and the admixture P1P2 (5.33). The value of the He statistic in subpopulation P2 (0.779) exhibited a slight increase compared to subpopulations P1 (0.779) and P1P2 (0.779). In this study, the obtained He values support the likelihood of outbreeding by approximately 78% during seed propagation (Table 2).
Genetic diversity and phylogenetic analysis
This study presents a comprehensive examination of rice genotypes from Egypt, employing a structural analysis approach. The investigation focuses on two distinct subpopulations, namely P1 and P2, and utilizes a set of 23 microsatellite markers for the analysis. A neighbor-joining tree without weighting was also constructed to verify the genetic relationships; it was accomplished using the Darwin software program (see Fig. 3). According to the findings derived from the STRUCTURE analysis, the germplasm accessions were categorized into two distinct clusters: blue (consisting of 12 genotypes) and red (comprising 15 genotypes). Therefore, the phylogenetic tree analysis further validates the STRUCTURE analysis, as it reveals the presence of two distinct clusters consistent with the previous findings.

Unrooted neighbor-joining tree of the 27 rice genotypes and based on data of 23 microsatellite markers. Cluster I is shown in blue color and cluster II is shown in red color. Bootstrap values are present in the middle of the branches. The numbers present at the tip of the branches indicate the number of genotypes
The analysis of molecular variance (AMOVA) using microsatellite data was calculated using ARLEQUIN version 3.1, considering both within and among subpopulations. Table 3 displays the results of the analysis of molecular variance (AMOVA) conducted using 23 microsatellite markers. The allelic distance matrix was utilized as input for the calculation of F-statistics. According to Table 3, the molecular variance percentage for the two subpopulations suggests that 34.22% of the variation can be attributed to differences between populations, while 65.78% can be attributed to differences within the population. It is anticipated that there will be no individual variation within single populations. A statistically significant fixation index (FST) value of 0.342 was observed (P > 0.001), along with a gene flow (Nm) value of 0.481.
The genotypes were clustered using neighbor-joining and structure-based populations, and it was observed that they exhibited a comparable pattern. Consequently, further investigation was conducted to examine their co-linearity utilizing the Venn diagram. The Venn diagram depicting the analysis of 27 genotypes revealed the presence of neighboring-joining clusters and model-based populations. The results indicated that 37% of the genotypes obtained from POPI exhibited an identical match with cluster I. Similarly, a significant proportion of genotypes (55.6%) from the POPII dataset exhibited similarity with cluster II, as depicted in Fig. 4. Nevertheless, it is worth noting that a mere 7.4% of genotypes derived from the POPI dataset exhibited complete similarity to cluster II. In general, the analysis revealed a notable resemblance between cluster grouping and Structure-based subpopulations as depicted in the Venn diagram, thereby enhancing the validity of the findings in the current investigation.

Estimation of co-linearity between neighbor-joining-based clusters and model-based sub-populations using Venn diagram. The total number of identical germplasms between cluster and sub-population is shown in the figure. The percentage of co-linearity is reported in parentheses
Principle component analysis (PCA)
Principal Component Analysis (PCA) demonstrated the interrelationship and grouping of the examined genotypes (old and modern groups) based on molecular data. According to the data presented in Fig. 5, it can be observed that PCA1 explains 17.4% of the total variance, whereas PCA2 accounts for a comparatively lower proportion of 10.2%. The principal component analysis (PCA) results revealed that the genotypes of the modern group were situated between those of the old group. According to the findings of the principal component analysis, a total of 27 components were identified. However, only the first four components, namely PC1, PC2, PC3, and PC4, were utilized to represent the variation. According to Table 4, the initial four components exhibited the most significant eigenvalues, specifically 18.413, 10.759, 7.618, and 7.031, respectively. The initial principal component, PC1, accounted for 17.37% of the overall variability. Principal Component 2 (PC2) accounted for 10.15% of the total variation, while Principal Component 3 (PC3) collectively accounted for 7.18%. Furthermore, the combined effect of PC3 explained 6.633% of the total variation.

Principle component analysis of the studied genotypes (old and modern groups) based on the molecular attributes
Discussion
Understanding and examining genetic diversity and the evolutionary interconnection among genotypes are essential in breeding and enhancement initiatives for strategic fields such as rice. Rice exhibits a restricted genetic background due to the frequent utilization of shared parents despite globally active breeding, genetic enhancement initiatives, and diverse genotypes (Bharadwaj et al. 2013; Safhi et al. 2022; Ibrahim et al. 2023a, b). Hence, it is imperative to consistently assess the genetic diversity of germplasm in order to ascertain the specific alleles that contribute to enhanced productivity and resilience against diverse environmental pressures, encompassing both biotic and abiotic stresses. The assessment of genetic diversity serves as a crucial means for the introduction of novel genetic material. The process of selection and hybridization offers valuable insights to plant breeders, enabling them to develop novel and versatile cultivars; its practice broadens rice's genetic diversity (Glaszmann et al. 2010; Essa et al. 2023, Mesfer ALshamrani et al. 2022; Essa et al. 2023).
The present investigation demonstrated the novel contributions of rice genotypes to comprehend their genetic diversity, population structure, and gene flow, aiming to facilitate breeding and enhance crop improvement efforts. Plant genetic resources (PGRs) are crucial in breeding and crop enhancement initiatives. However, it is important to note that these Plant Genetic Resources (PGRs) are considered the most exemplary representatives of a wide range of genetic material. Nonetheless, it is crucial to possess prior research and understanding of genetic diversity to effectively utilize these PGRs (Bueno et al. 2019; Safhi et al. 2022; Soliman et al. 2023). Over the past few decades, there has been notable advancement in molecular marker methods, which have proven effective in assessing the extensive genetic diversity of germplasm resources (Mesfer ALshamrani et al. 2022; Naaz et al. 2022). Based on prior molecular marker data, genetic diversity was assessed using a panel of 23 microsatellite markers. All of the samples exhibited polymorphisms, indicating genetic variation, and were consequently employed in investigating genetic diversity. About 106 alleles were observed across 23 genotypes, averaging 4.61 per locus. Previous research also observed comparable outcomes in rice by examining microsatellites (Ahmed et al. 2019; Naaz et al. 2022; Hassan and Hama-Ali 2022).
Botstein et al. (1980) state that a marker with a polymorphic information content (PIC) value greater than or equal to 0.5 is considered highly informative. Conversely, a marker with a PIC value below 0.5 is classified as moderately informative, while a marker with a PIC value below 0.25 is categorized as minimally informative. The study observed a range of PIC values for the 23 microsatellite loci, from 0.358 to 0.751. The average PIC value was calculated to be 0.580, suggesting that these markers possess a high level of informativeness. The usefulness of microsatellite loci and their characteristics for detecting genotype differences is evaluated by PIC (Maclean et al. 2013; EL-Mansy et al. 2021; Abd El-Moneim et al. 2021; Essa et al. 2023). Consequently, the calculated average PIC value of approximately 0.6 indicates that the rice genotypes employed in this investigation exhibited substantial genetic diversity. Out of the total 23 microsatellite loci examined, it was observed that five specific loci (RM5, RM11, RM19, RM161, RM413, and RM474) exhibited notably high polymorphic information content (PIC) values, ranging from 0.703 to 0.751. Including these highly polymorphic microsatellite loci can potentially enhance the genetic diversity of existing genotypes. The utilization of these high-density microsatellite loci has the potential to expand the genetic diversity of current genotypes. Singh et al. (2013) and Naaz et al. (2022) reported a similar average PIC value. The determination of PIC (Polymorphic Information Content) values is subject to the influence of several parameters, such as the breeding method employed by the species, the level of genetic variability present in the selected genotypes, the size of the population under study, the specific genotypic technique utilized, and the location of primer sites within the genome (Singh et al. 2013; Naaz et al. 2022). The current genotypes exhibit a wide range of genetic diversity, spanning from 0.358 to 0.751, with an average value of 0.580. The findings of this study align with the results reported by Babu et al. (2014) in their investigation of 82 different rice genotypes. Previous research estimated the PIC values as follows: 0.240 (Anandan et al. 2016), 0.416 (Nachimuthu et al. 2015), 0.560 (Pathaichindachote et al. 2019), 0.630 (Aljumaili et al. 2018), and 0.738 (Wang et al. 2014).
The microsatellite loci exhibited considerable variation in the number of alleles, ranging from 2 alleles for four loci (RM144, RM151, RM215, and RM277) to 8 alleles for RM11, with an average of 4.61 alleles per locus. Thomson et al. (2009) reported comparable results. Maclean et al. (2013) documented the count of unique alleles for 11 microsatellite loci (RM5, RM55, RM118, RM133, RM154, RM215, RM271, RM277, RM413, RM433, and RM474) within a set of 82 rice genotypes. The analysis shows that three loci (RM133, 154, and 271) exhibited an equivalent count of unique alleles (Babu et al. 2014). The study revealed a significant and robust correlation between the number of alleles and both polymorphic information content (PIC) (r = 0.785**) and gene diversity (r = 0.713**). This finding presents an additional explanation for the notable genetic variability observed among the 27 rice genotypes. Therefore, these microsatellite loci show high productivity and can be effectively utilized to investigate genetic variation.
This study utilized 106 microsatellite alleles to assess the population structure of 27 Egyptian rice genotypes. The analysis was conducted using 23 microsatellite loci. Prior research established microsatellites as DNA markers in plants exhibiting substantial polymorphism (Naaz et al. 2022; Hassan and Hama-Ali 2022). At the highest delta K value, 27 rice genotypes were segregated into two distinct subpopulations, P1 and P2. These subpopulations exhibited significant admixture variations, contributing to a considerable variance. Singh et al. (2013) reached comparable findings. The AMOVA study revealed a statistically significant level of genetic variability within the examined population, amounting to 65.78%. The level of genetic diversity observed among subpopulations was minimal, specifically measuring 34.22%. The observed genetic variation among different genotypes can potentially be ascribed to the process of gene flow, which is facilitated by the mobility of seeds (Dhanapal et al. 2015). The practice of seed exchange among farmers contributes to the augmentation of local germplasm diversity. Hence, using model-based population structure analysis facilitates the identification and mapping of significant agronomical traits while also enabling the examination of recombination patterns through genome-wide association analysis.
Consequently, the dispersion of alleles across different populations exhibits an increase that is not influenced by their geographical proximity (Louette et al. 1997). Choudhary et al. (2013) observed a substantial evolutionary divergence among five rice populations, amounting to 92.12%. Conversely, the genetic diversity within each population was comparatively low, accounting for only 7.88%. The distribution of genetic diversity among 375 rice genotypes was observed as follows: 4% among populations consisting of 12 distinct populations, 70% among individuals, 25% within individuals, and 1% among regions (Singh et al. 2013). The current investigation demonstrates that the considerable degree of polymorphism observed among genotypes in P1 and P2 possesses the potential to enhance genetic diversity in breeding initiatives (Thomson et al. 2009).
In AMOVA analysis, FST is a metric for quantifying population differentiation by evaluating genetic structure. Wright (1965) significantly contributed to genetics by identifying genetic differentiation utilizing FST values. The three levels of differentiation, as defined by previous research (Wright 1978), are as follows: low differentiation (FST = 0.00–0.05), moderate differentiation (FST = 0.05–0.15), and high differentiation (FST > 0.30). According to the study conducted by Frankham et al. (2010), a population can be considered significantly differentiated when the FST value exceeds 0.15. Within the present context, a notable disparity was observed in the FST values (0.342) between the two subpopulations. Verma et al. (2019), Gouda et al. (2020), and Suvi et al. (2020) reported significant genetic differentiation (FST) values of 0.827, 0.490, and 0.407, respectively, when examining subpopulations. In addition, Nm is a valuable tool for assessing the significance of gene flow and genetic drift in evolutionary differentiation (Hassan and Hama-Ali 2022). Gene flow between populations within a species maintains genetic diversity, and a higher frequency of gene flow contributes to increased genetic variation (Slatkin 1994). In contrast, gene flow diminishes genetic differentiation as genetic diversity increases. Consequently, the augmentation of gene flow is expected to lead to a decline in genetic differentiation, thereby enhancing genetic diversity (Fu et al. 2016). The Nm value (0.481) observed in our study is significantly lower than the critical value of 1, suggesting a substantial genetic divergence between the two subpopulations (P1 and P2). According to Slatkin (1985), a value of Nm less than one signifies a restricted gene flow between populations.
The results of the neighbor-joining tree analysis exhibited congruent patterns concerning population structure. The classification of all 27 genotypes into two distinct groups with similar genotypes in the evolutionary tree was exemplified by the findings of the STRUCTURE analysis. The Unweighted neighbor-joining tree is utilized to depict the level of genetic diversity among different rice genotypes. Moreover, the study on co-linearity uncovered the presence of cluster grouping and subpopulations based on structural characteristics. The subpopulation P1 in the STRUCTURE analysis exhibits a similarity of 37% with cluster I in the dendrogram.
Similarly, the subpopulation denoted as P2 in the STRUCTURE analysis exhibits a 55.6% similarity with cluster II. The previous study provides further evidence supporting the results obtained from cluster and structure-based population analyses. Additionally, it highlights the efficacy of microsatellite-based genotyping as a reliable approach for investigating genetic variation in rice.
Furthermore, the population structure analysis revealed the presence of additional admixtures. The structural analysis revealed the presence of admixtures (specifically genotypes 16, 17, and 21) that are distinctly positioned in the dendrogram. This observation underscores the significant possibility of gene flow occurring between these genotypes, even in the presence of geographic barriers. Gene flow may manifest itself through the exchange of seeds for cultivation purposes among growers. Consequently, utilizing the model-based population structure analysis in the present study may contribute to mapping important agronomical traits and examining recombination patterns through genome-wide association analysis.
A unique allelic pattern was observed in each subpopulation. The variability of Na and Ne was detected to be somewhat diverse in both groups. The absence of heterozygosity was reflected in all microsatellite loci due to the high degree of homozygosity in the genotypes. Moreover, due to the self-pollination nature of rice, it is expected that microsatellite markers would exhibit only a single allele at each locus. Consequently, the level of heterozygosity is expected to be null. Firstly, the variability of Na and Ne (allele number and effective allele number) being somewhat diverse in both groups suggests that different alleles are present within each subpopulation. This could result from genetic mutations, recombination events, or natural selection acting on different alleles in each subpopulation. The absence of heterozygosity, meaning that both alleles at each microsatellite locus are the same, can be explained by the high degree of homozygosity in the genotypes (Yun et al. 2020). Homozygosity is the condition where an individual carries two identical alleles for a particular gene or genetic marker, and it can arise due to self-pollination. In the case of rice, which has a self-pollination nature, it is expected that the microsatellite markers would exhibit only a single allele at each locus, resulting in a lack of heterozygosity. Consequently, the level of heterozygosity is expected to be null because of the specific reproductive system of rice and the resulting high degree of homozygosity. This means that within each subpopulation, individuals are likely to carry the same two alleles for each microsatellite locus, leading to a unique allelic pattern in each group (Uddin et al. 2022).
Nevertheless, a substantial range was observed in the expected heterozygosity values, from 0.475 to 0.794. Measuring heterozygosity (He) in population genetics is a fundamental parameter that provides valuable insights into genetic diversity (Yu et al. 2013). The systemic inflammatory response (SII) was detected in the analysis of all 23 microsatellite loci. The SII values, ranging from 0.659 to 1.690, observed across all microsatellite loci suggest that all loci exhibit informativeness due to utilizing a genetically diverse population in the current investigation. Furthermore, it was observed that SII and Ne showed a significant and robust positive correlation (r = 0.941**).
Subpopulation P1 possesses genotypes originating from four distinct released groups, rendering it a highly valuable reservoir of genetic diversity in rice. Utilizing these genotypes will prove advantageous in future breeding initiatives that augment rice's genetic diversity. Multi-parent advanced generation inter-crosses (MAGIC) can potentially benefit marker-assisted selection and genome-wide association studies due to their enhanced diversity. The primary sources of information utilized for addressing the issue of limited gene flow in rice imports within the region were the outcomes of AMOVA, FST, and Nm analyses. It is conceivable that a substantial proportion of genotypes within the population exhibit a high degree of similarity, and it is also plausible that the practice of seed sharing among farmers is not widespread. Farmers do not receive seeds from governmental entities. Consequently, farmers reserve a portion of their seeds annually for sowing in the subsequent year. Principal Component Analysis (PCA) is a widely used unsupervised learning technique employed to decrease the dimensionality of a given dataset and extract the most significant information for subsequent analysis (Iqbal et al. 2014).
Conclusions
The results suggest that microsatellite markers are valuable tools for assessing genetic diversity among rice cultivars. The process of selecting genotypes as potential donors in a crop breeding strategy aimed at enhancing specific attributes in rice necessitates the examination of genetic diversity among individuals within populations. Moreover, it is crucial to comprehend the distribution of alleles within subpopulations as it provides insights into the valuable loci that can be employed to investigate genetic diversity. The resolution of the issue concerning inadequate gene flow in rice imports in the region relied heavily on utilizing AMOVA, FST, and Nm findings as primary sources of information.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Abd El-Moneim D, ELsarag EIS, Aloufi S, El-Azraq AM, Alshamrani SM, Safhi FAA, Ibrahim AA (2021) Quinoa (Chenopodium quinoa Willd.): genetic diversity according to ISSR and SCoT markers, relative gene expression, and morpho-physiological variation under salinity stress. Plants 10:2802. https://doi.org/10.3390/plants10122802
Ahmed S, Anik TR, Islam A, Uddin I, Haque MS (2019) Screening of some rice (Oryza sativa L.) genotypes for salinity tolerance using morphological and molecular markers. Biosci Biotech Res Asia 16(2):377–390
Aljumaili SJ, Rafii M, Latif M, Sakimin SZ, Arolu IW, Miah G (2018) Genetic diversity of aromatic rice germplasm revealed by SSR markers. Biomed Res Int 25:7658032
Anandan A, Anumalla M, Pradhan SK, Ali J (2016) Population structure, diversity and trait association analysis in rice (Oryza sativa L.) germplasm for early seedling vigor (ESV) using trait linked SSR markers. PLoS ONE 11(3):e0152406
Babu BK, Meena V, Agarwal V, Agrawal PK (2014) Population structure and genetic diversity analysis of Indian and exotic rice (Oryza sativa L.) accessions using SSR markers. Mol Biol Rep 41(7):4329–4339
Bharadwaj C, Srivastava R, Chauhan SK, Satyavathi CT, Kumar J, Faruqui A, Yadav S, Rizvi AH, Kumar T (2013) Molecular diversity and phylogeny in geographical collection of chickpea (Cicer sp.) accessions. J Genet 92:94–100
Borba TCO, Brondanil RPV, Breseghello F, Coelho ASG, Mendonça JA, Range PHN, Brondani C (2010) Association mapping for yield and grain quality traits in rice (Oryza sativa L.). Gen Mol Biol 33:515–524
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Bueno E, Kisha T, Maki SL, Von Wettberg EJB, Singer S (2019) Genetic diversity of Chamaecrista fasciculata (Fabaceae) from the USDA germplasm collection. BMC Res Notes 12(117):1–7
Choudhary G, Ranjitkumar N, Surapaneni M, Deborah DA, Vipparla A, Anuradha G, Siddiq EA, Vemireddy LR (2013) Molecular genetic diversity of major Indian rice cultivars over decadal periods. PLoS ONE 8(6):e66197
Dhanapal AP, Ray JD, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, Andy King C, Cregan PB, Song Q, Fritschi FB (2015) Genome-wide association study (GWAS) of carbon isotope ratio (δ13C) in diverse soybean [Glycine max (L.) Merr.] genotypes. Theor Appl Genet 128(1):73–91
Earl DA, Von Holdt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
EL-Mansy AB, Aoneim D, AL-Shamrani SM, Alsafhi FA, Abdein MA, Ibrahim AA (2021) Genetic diversity analysis of tomato (Solanum lycopersicum L.) with morphological, cytological, and molecular markers under heat stress. Horticulturae 7:65
Essa NM, Ibrahim AA, Soliman MI (2023) Association studies between some cultivated species and their wild relatives from Apiaceae, Asteraceae and Brassicaceae families based on molecular and DNA barcoding in Egypt. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-023-01681-x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587
Frankham R, Ballou JD, Briscoe DA (2010) Introduction to conservation genetics, 2nd edn. Cambridge University Press, Cambridge, pp 309–328
Fu PC, Gao QB, Zhang FQ, Xing R, Khan G, Wang JL, Liu HR, Chen SL (2016) Responses of plants to changes in Qinghai-Tibetan Plateau and glaciations: evidence from phylogeography of a Sibiraea (Rosaceae) complex. Biochem Syst Ecol 65:72–82
Glaszmann JC, Kilian B, Upadhyaya HD, Varshney RK (2010) Accessing genetic diversity for crop improvement. Curr Opin Plant Biol 13(2):167–173
Gouda AC, Ndjiondjop MN, Djedatin GL, Warburton ML, Goungoulou A, Kpeki SB, N’Diaye A, Semagn K (2020) Comparisons of sampling methods for assessing intra-and inter-accession genetic diversity in three rice species using genotyping by sequencing. Sci Rep 10(1):1–14
Hassan DA, Hama-Ali EO (2022) Evaluation of gene flow and genetic diversity in rice accessions across Kurdistan region-Iraq using SSR markers. Mol Biol Rep 49:1007–1016
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9(5):1322–1332
Ibrahim AA, Alwutayd KM, Safhi FA et al (2023a) Characterization and comparative genomic analyses of complete chloroplast genome on Trema orientalis L. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-023-01678-6
Ibrahim AA, Salem KFM, Abdein MA, Ramadan SM (2023b) Advances in summer squash (Cucurbita pepo L.) molecular breeding strategies. In: Singh S, Sharma D, Sharma SK, Singh R (eds) Smart plant breeding for vegetable crops in post-genomics era. Springer, Singapore. https://doi.org/10.1007/978-981-19-5367-5_8
Iqbal Q, Saleem MY, Hameed A, Asghar M (2014) Assessment of genetic divergence in tomato through agglomerative hierarchical clustering and principal component analysis. Pak J Bot 46(5):1865–1870
Khush GS (2013) Strategies for increasing the yield potential of cereals: case of rice as an example. Plant Breed 132(5):433–436
Louette D, Charrier A, Berthaud J (1997) In situ conservation of maize in Mexico: genetic diversity and maize seed management in a traditional community. Econ Bot 51(1):20–38
Lui K (2005) Power marker: integrated analysis environment for genetic marker data. Bioinform 21:2128–2129
Maclean J, Hardy B, Hettel G (2013) Rice almanac, fourth ed. International Rice Research Institute. http://irri.org/resources/publications/books/rice-almanac-4th-edition.
Mesfer AL-Shamrani S, Safhi FA, Alshaya DS, Ibrahim AA, Mansour H, Abd El Moneim D (2022) Genetic diversity using biochemical, physiological, karyological and molecular markers of Sesamum indicum L. Front Genet 13:1035977. https://doi.org/10.3389/fgene.2022.1035977
Mohanty S, Donde R, Das S, Panda D, Mishra B, Pradhan SK, Dash SK, Swain P, Behera L (2021) Utilization of genetic diversity and population structure to reveal prospective drought-tolerant donors in rice. Gene Rep 23:101151
Mourad AMI, Alomari DZ, Alqudah AM, Sallam A, Salem KFM (2019) Recent advances in wheat (Triticum spp.) breeding. In: Al-Khayri J, Jain S, Johnson D (eds) Advances in plant breeding strategies: cereals. Springer, Cham. https://doi.org/10.1007/978-3-030-23108-8_15
Naaz S, Pandey V, Yadav HK (2022) Evaluation of genetic diversity in rice (Oryza sativa L. ssp. Indica) accessions using SSR marker. Vegetos. https://doi.org/10.1007/s42535-022-00378-3
Nachimuthu VV, Muthurajan R, Duraialaguraja S, Sivakami R, Pandian BA, Ponniah G, Gunasekaran K, Swaminathan M, Suji K, Sabariappan R (2015) Analysis of population structure and genetic diversity in rice germplasm using SSR markers: an initiative towards association mapping of agronomic traits in Oryza sativa. Rice 8(1):1–25
Oliveros JC (2007) Venny. An interactive tool for comparing lists with venn diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html
Pathaichindachote W, Panyawut N, Sikaewtung K, Patarapuwadol S, Muangprom A (2019) Genetic diversity and allelic frequency of selected Thai and exotic rice germplasm using SSR markers. Rice Sci 26(6):393–403
Perrier X, Jacquemoud-Collet JP (2006) Darwin software http://darwin.cirad.fr/Darwin
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rai GK, Khanday DM, Kumar P, Magotra I, Choudhary SM, Kosser R, Kalunke R, Giordano M, Corrado G, Rouphael Y et al (2023) Enhancing crop resilience to drought stress through CRISPR-Cas9 genome editing. Plants 12:2306. https://doi.org/10.3390/plants12122306
Safhi FA, Alshamrani SM, Jalal AS, El-Moneim DA, Alyamani AA, Ibrahim AA (2022) Genetic characterization of some Saudi Arabia’s accessions from Commiphora gileadensis using physio-biochemical parameters, molecular markers, DNA barcoding analysis and relative gene expression. Genes 13:2099. https://doi.org/10.3390/genes13112099
Salem KFM (2004) The inheritance and molecular mapping of genes for post-anthesis drought tolerance (PADT) in wheat. Doctoral dissertation, Ph. D. Dissertation, Martin Luther University, Halle-Wittenberg, Germany
Singh N, Choudhury DR, Singh AK, Kumar S, Srinivasan K, Tyagi RK, Singh NK, Singh R (2013) Comparison of SSR and SNP markers in estimation of genetic diversity and population structure of Indian rice varieties. PLoS ONE 8(12):1–14
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39(1):53–65
Slatkin M (1994) Gene flow and population structure. In: Real LA, Princeton NJ (eds) Ecological genetics. Princeton University Press, pp 3–17
Soliman MI, Mohammed NS, Safhi FA, AL-Shamrani SM, Alyamani AA, Alharthi B, Qahl SH, Al Kashgry NA, Ibrahim AA (2023) Antibacterial, antioxidant activities, GC-mass characterization, and cyto/genotoxicity effect of green synthesis of silver nanoparticles using latex of Cynanchum acutum L. Plants 12(1):172. https://doi.org/10.3390/plants12010172
Suvi WT, Shimelis H, Laing M, Mathew I, Shayanowako AIT (2020) Assessment of the genetic diversity and population structure of rice genotypes using SSR markers. Acta Agric Scand B 70(1):76–86
Thomson MJ, Polato NR, Prasetiyono J, Trijatmiko KR, Silitonga TS, McCouch SR (2009) Genetic diversity of isolated populations of Indonesian landraces of rice (Oryza sativa L.) collected in East Kalimantan on the island of Borneo. Rice 2(1):80–92
Uddin N, Muhammad N, Ali SS, Ullah R, Bari A, Hussain H, Zhu D (2022) Characterization of the genetic variability within Ziziphus nummularia genotypes by phenotypic traits and SSR markers with special reference to geographic distribution. Genes. https://doi.org/10.3390/genes14010155
Verma H, Borah JL, Sarma RN (2019) Variability assessment for root and drought tolerance traits and genetic diversity analysis of rice germplasm using SSR markers. Sci Rep 9(1):16513
Wang C, Zheng X, Xu Q, Yuan X, Huang L, Zhou H, Wei X, Ge S (2014) Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity 112(5):489–496
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420
Wright S (1978) Evolution and the genetics of populations: a treatise in four volumes: variability within and among natural populations, vol 4. University of Chicago Press, Chicago
Wu J, Tanksley SD (1993) Abundance, polymorphism and genetic mapping of microsatellites in rice. Mol Gen Genet 241(1):225–235
Yeh FC, Yang RC, Boyle TBJ, Ye Z, Mao J (1997) POPGENE: the user-friendly shareware for population genetics analysis. Mol Biol Biotech Cent 10:295–301
Yu J, Zhao H, Zhu T, Chen L, Peng J (2013) Transferability of rice SSR markers to Miscanthus sinensis, a potential biofuel crop. Euphytica 191(3):455–468
Yun SA, Son HD, Im HT, Kim SC (2020) Genetic diversity and population structure of the endangered orchid Pelatantheria scolopendrifolia (Orchidaceae) in Korea. PLoS ONE 15(8):e0237546. https://doi.org/10.1371/journal.pone.0237546
Acknowledgements
Co-author K.F.M.S. would like to thank the Deanship of Scientific Research at Shaqra University, Saudi Arabia for supporting this research work. Co-author A.A.I. would like to thank the Botany and Microbiology Department, Faculty of Science, Arish University, Egypt for scientific support; also all authors thank Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R318), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Funding
Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R318), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, KFMS, ASMR and AAI; methodology, KFMS and AAI; software, AAI & PG & KFMS; validation, KFMS, ASMR & AAI; formal analysis, KFMS and AAI; investigation, KFMS and AAI; resources, KFMS, ASMR & AAI; data curation, KFMS AAI & PG; writing—review and editing, all authors; supervision, KFMS; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Salem, K.F.M., Safhi, F.A., Alwutayd, K.M. et al. Analysis of genetic diversity, population structure and phylogenetic relationships of rice (Oryza sativa L.) cultivars using simple sequence repeat (SSR) markers. Genet Resour Crop Evol 71, 2213–2227 (2024). https://doi.org/10.1007/s10722-023-01789-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-023-01789-0