
Summary
Background This Phase-I-study aimed to determine the recommended Phase-II-dosing-schedule of LY2334737, an oral gemcitabine prodrug, in patients with advanced/metastatic solid tumors. Pharmacokinetics, cytokeratin-18 (CK18) levels, genetic polymorphisms, and antitumor activity were additionally evaluated. Methods Patients received escalating doses of LY2334737 either every other day for 21 days (d) followed by 7 days-drug-free period (QoD-arm) or once daily for 7 days every other week (QD-arm). The 28 days-cycles were repeated until disease progression or unacceptable toxicity. Standard 3 + 3 dose-escalation was succeeded by a dose-confirmation phase (12 additional patients to be enrolled on the maximum tolerated dose [MTD]). Results Forty-one patients received QoD- (40–100 mg) and 32 QD-dosing (40-90 mg). On QoD, 3/9 patients experienced dose-limiting toxicities (DLTs) on the 100 mg dose (2 × G3 diarrhea, 1 × G3 transaminase increase); 1 additional DLT (G3 diarrhea) occurred during dose confirmation at 90 mg (12 patients). On QD, 1 patient each experienced DLTs on 60 mg (G3 transaminase increase) and 80 mg (G3 prolonged QTcF-interval); 2/7 patients had 3 DLTs on the 90 mg dose (diarrhea, edema, liver-failure; all G3). The MTD was established at 90 mg for the QoD-arm. Seven patients on QoD and 4 on QD achieved SD (no CR + PR). Pharmacokinetics showed a dose-proportional increase in exposure of LY2334737 and dFdC without accumulation after repeated dosing. Significant increases in CK18 levels were observed. Genetic polymorphism of the cytidine deaminase gene (rs818202) could be associated with ≥ G3 hepatotoxicity. Conclusions Both schedules displayed linear pharmacokinetics and acceptable safety profiles. The recommended dose and schedule of LY2334737 for subsequent Phase-II-studies is 90 mg given QoD for 21 day.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gemcitabine (2’, 2’-difluorodeoxycytidine, dFdC) is approved for systemic chemotherapy in pancreatic, non-small-cell lung, ovary, bladder, and breast cancer [1]. Efficacy and toxicity of gemcitabine are known to highly depend on the schedule of administration [2–4]. Previous studies have demonstrated that long-term continuous exposure to low-dose gemcitabine could provide pharmacokinetic (PK) advantages by reducing the renal clearance of gemcitabine and optimizing the pharmacological phosphorylation of dFdC into dFdC triphosphate by cytidine kinase, a saturable enzyme that was considered as a limiting step for gemcitabine activity [2, 4–7]. An oral formulation sharing a similar mechanism of action with gemcitabine could allow achieving sustainable exposures and would limit the discomfort associated with the use of a protracted infusion device. However, dFdC has poor bioavailability if administered by oral route [8] due to an extensive first-pass metabolism by the cytidine deaminase (CDA) into its main metabolite 2’, 2’-difluorodeoxyuridine (dFdU) [9]. Several prodrugs and dFdC derivatives have been developed to protect against CDA, to improve intracellular uptake, and/or to prolong the release of dFdC in cancer cells [10]. LY2334737 was designed as a molecule where dFdC was covalently linked to valproic acid (VPA) by a carboxylesterase-sensitive linker [11]. When given orally, LY2334737 can be absorbed in the gut and is then hydrolyzed by carboxylesterase II (CES2), releasing dFdC and VPA into the systemic circulation [12].
The first-in-human Phase I study of LY2334737 in European patients explored a once daily (QD) for 14 days schedule, with or without erlotinib, followed by 7 drug-free days [13]. The maximum tolerated dose (MTD) was defined as 40 mg/day. Another Phase I study performed in Asian patients showed unexpected hepatic toxicities [14]. These findings indicated that alternative dosing schedules might be required.
Therefore, this Phase I study of single-agent LY2334737 evaluated 2 alternative schedules based on 28-day cycles. Patients with advanced and/or metastatic solid tumors received LY2334737 either every other day (QoD) for 21 days, followed by 7 drug-free days (QoD arm), or once daily for 7 days every other week (QD arm). The aim was to determine the recommended dose of LY2334737 for subsequent Phase II studies. Genotyping was performed to assess CDA and other germline polymorphisms that might potentially be associated with hepatic toxicity [14]. PK of LY2334737, dFdC, and dFdU, along with pharmacodynamics data looking at DNA incorporation of dFdC in peripheral blood mononuclear cells were also assessed.
Materials and methods
Study design and patients
This Phase I dose escalation trial (I1C-MC-JLBE) evaluated 2 schedules of oral LY2334737 in parallel. Key eligibility criteria included pathologically or cytologically proven advanced and/or metastatic solid tumor, no standard therapeutic option, age ≥18 years, performance status 0–2 (Eastern Cooperative Oncology Group [ECOG]), estimated life expectancy ≥12 weeks. Previous cancer treatments had to be discontinued for at least 30 days before study entry. Patients had to have adequate bone marrow function (hemoglobin ≥9.0 g/dL, neutrophil count ≥1.5 × 109/L, platelet count ≥100 × 109/L), adequate renal function (serum creatinine ≤1.5× the upper limit of normal [ULN]), and adequate hepatic function (aspartate transaminase [AST] and alanine transaminase [ALT] ≤2.5 × ULN, bilirubin ≤1.5 × ULN). Patients with liver cirrhosis, chronic hepatitis, history of excessive alcohol consumption, and patients positive for hepatitis B antigen, hepatitis C virus, or human immunodeficiency virus antibodies were excluded. Patients with gastrointestinal disease that might interfere with oral study drug absorption and patients with a history of severe hypersensitivity to dFdC were also excluded. Concomitant chemotherapy, radiotherapy, immunotherapy, or hormonal therapy for cancer, and VPA treatment were prohibited.
Patients who provided written informed consent were recruited at 4 different sites in France (2), Germany (1), and the United States (1) between September 2008 and November 2012. The study was performed according to the Declaration of Helsinki and approved by the responsible ethical review boards in each country.
Study treatment
Eligible patients were successively assigned to escalating doses of LY2334737, using 1 of the 2 different schedules based on 28-day cycles. Patients enrolled into the QoD arm took LY2334737 every other day for 21 days, followed by 7 drug-free days. Patients enrolled into the QD arm received repeated sequences of LY2334737 QD for 7 days followed by 7 drug-free days. Each 28-day-cycle was repeated until disease progression or unacceptable toxicity.
LY2334737 was supplied as 5, 15, and 30 mg capsules in blister packs for oral administration. During Cycles 1 and 2, patients were instructed to take LY2334737 with a glass of water approximately 30 min prior to the morning meal, at the same time every day. During Cycles 3 and beyond, patients were given the option of taking LY2334737 in the evening (approximately 30 min prior to the evening meal) if the treating physician thought it would improve treatment tolerability.
Dose escalation and dose confirmation
A conventional 3 + 3 design was used for dose escalation of LY2334737; intra-patient dose escalation was not allowed. The 3 initial patients in both arms (QoD and QD) received 40 mg. Doses were escalated based on the occurrence of dose-limiting toxicities during Cycle 1 (DLTs). Safety data were the primary driver for the dose escalation and the PK data were used as additional supporting data. Based on the safety information obtained from a previous study [13], the dose escalation was initially restricted to a maximum of 25 % per dose level for both dosing regimen.
Once the MTD had been defined, up to 12 additional patients were to be enrolled to confirm feasibility of the dose selected. Based upon safety and PK considerations, dose confirmation could be opened for either both, or only 1 of the 2 dosing regimens. The rationale for expanding the MTD cohort by 12 patients was to confirm the MTD, and to enroll a sufficient number of patients to enable exploratory pharmacodynamic and biomarker analyses.
DLT and MTD
A DLT was defined as occurrence of any of the following events during Cycle 1: Non-hematological toxicity ≥ Grade (G) 3 other than nausea/vomiting, G3 neutropenia with fever or G4 neutropenia, G3 thrombocytopenia associated with ≥ G2 bleeding or G4 thrombocytopenia, >14 days needed to recover from toxicity after the last dose of Cycle 1, any other significant drug-related toxicity considered to be dose-limiting by the investigator. The MTD was defined as the highest dose of LY2334737 that showed DLTs in no more than 2 of 6 patients (i.e., 33 % probability of causing a DLT in a specific cohort).
Safety evaluations
Patients were monitored for safety on a weekly basis. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 15.1. Standard laboratory tests were performed to monitor safety, and laboratory toxicities were graded using the Common Terminology Criteria for Adverse Events (CTC-AE) Version 3.0. Twelve-lead electrocardiograms (ECGs) were obtained pre-study, on Days 1 and 21 of Cycle 1 at 3 different time points (30 min pre-dose, 2 h, and 7 h post-dose), and reviewed for any clinically significant ECG changes. Triplicate QT interval data were averaged for each period and at each time point. The ECGs measured during Day -1, together with the pre-dose ECG on Day 1 of Cycle 1, were considered the “baseline” ECGs. Changes in QT from baseline were calculated by subtracting the respective reading taken at the same nominal time on Day -1 from the reading taken after LY2334737 dosing on Days 1 and 21.
Tumor response
No formal efficacy analysis was performed, but lesion and response data were reported following Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 criteria [15].
Pharmacokinetics
Plasma concentrations of LY2334737 and its metabolites dFdC and dFdU were assessed after single dose administration (Cycle 1 Day 1) and at steady state (Cycle 1 Day 7 and Day 21). On the respective days, 4 mL venous blood samples were collected at 1, 2, 4, 8, and 24 h after administration. All samples were drawn in sodium-heparinized tubes containing 100 μg/mL tetrahydrouridine to prevent deamination of dFdC into dFdU. Plasma concentrations were assayed at PharmaNet USA, Inc. (formerly Taylor Technology Inc.), Princeton, New Jersey, USA, using a validated high-pressure liquid chromatography-mass spectrometry/mass spectrometry method (HPLC-MS/MS). Plasma samples were extracted on Oasis HLB cartridges and separated on a Betasil C18 HPLC column with tandem mass spectrometric detection using positive ion atmospheric pressure chemical ionization. The lower limit of quantification was 0.1 ng/mL for LY2334737, 0.25 ng/mL for dFdC, and 1 ng/mL for dFdU.
Plasma PK parameters were analyzed by standard non-compartmental methods using the WinNonlin Professional Edition.
Incorporation of dFdC into DNA
Additional 10 mL blood samples were drawn into ethylene diamine tetraacetic acid (EDTA) tubes for exploratory analyses of dFdC incorporated into DNA. These samples were collected before the first intake of LY2334737 (≤7 days before start of Cycle 1), 24 h after the first dose, pre-dose on Days 7 and 13 of Cycle 1, 24 h after the Day 21 dose of Cycle 1, and pre-dose on Days 1, 7, and 21 of Cycle 2. The amount of dFdC and deoxyguanosine (dG) in DNA extracted from whole blood was determined by a validated liquid chromatography mass spectrometry [16]. The concentration was reported as the ratio of picograms of dFdC to micrograms of dG. Quantification of dFdG and dG were done by Advion BioSciences Inc, Ithaca, New York, USA.
Cytokeratin 18 M30 and M65 antigens
Additional 2 mL blood samples were drawn into EDTA tubes for evaluation of cytokeratin 18 (CK18) M30 and M65 antigen levels which are released from dying cells [17–19]. CK18 M30 and M65 antigen levels were determined by enzyme-linked immunosorbent assay (ELISA) [17].
Pharmacogenetic analyses
Anonymized DNA samples were collected from patients of those studies who had agreed to DNA sample banking and signed a separate informed consent. Seven single-nucleotide polymorphisms (SNPs) on 5 genes were selected for genotyping because they had been significantly associated with safety events in Asian LY2334737-treated patients [14] by previous genotyping [ILS Genomics, Morrisville, North Carolina, USA; data on file at Lilly]. These included 3 SNPs located in the human leukocyte antigen (HLA) gene complex: rs12526186 (C > T), rs2269706 (G > A), and rs3096691 (G > A). Additional SNPs genotyped on in this study include rs818202 (G > A) in the CDA gene, rs45523532 (G > T) in the SLC28A1 gene which encodes a gemcitabine transporter, rs2303218 (T > C) in the CES2 gene, and rs4148323 (G > A) in a glucuronidase gene involved in bilirubin physiology (UGT1A1).
TaqMan® genotyping assays and DNA sequencing (rs45523532 only) were used for SNP genotyping (details in Online Resource 1). SNPs with call rates <90 % were excluded from the study. SNP assessments with discordance rates >10 % in duplicate assays were also excluded. Allelic frequencies were compared versus the frequencies in the publically available HapMap CEU reference panel which consists of 30 parent-offspring trios from Utah with European ancestry (HapMap CEU cohort; www.hapmap.org). For the Caucasian subpopulation, the allelic distribution of SNPs was tested for Hardy-Weinberg equilibrium (HWE; see Online Resource 1).
Association of SNP variants with the following binary safety parameters was investigated for all patients and for Caucasians only: Level 2 and Level 3 hepatotoxicity (≥G2 and ≥ G3 increase of either AST, ALT, or bilirubin); ≥G1, ≥G2, and ≥ G3 increases of AST and ALT, and ≥ G2 increase of bilirubin, ≥G1 thrombocytopenia, and clustered AEs (≥G1 thrombocytopenia AND any occurrence of ≥ G3 increase of AST, ALT, or bilirubin). This was done by 2 × 2 contingency analyses using allelic, genotypic, dominant, and recessive statistical models and applying Fisher’s exact test [20]. Bonferroni multiplicity adjustment was performed within each endpoint. Associations with genetic variants with unadjusted p-values < 0.05 were considered of interest. In addition, box plots were created showing maximum AST, ALT, and bilirubin values against the different genetic variations of each SNP.
Post-hoc, an ENCODE analysis was performed by Bioinformatics on the SNP rs818202, to better understand the functional consequences of this SNP [21].
Results
Patient population
Seventy-three patients with a broad variety of advanced and/or metastatic solid tumors were enrolled (Table 1). Forty-one patients were enrolled into the QoD arm of the study, including 29 patients treated with escalating doses of 40-100 mg LY2334737, and 12 patients treated at the MTD for dose confirmation. Thirty-two patients were enrolled into the QD arm and received escalating doses of 40–90 mg LY2334737. Most patients (90.4 %) were Caucasian; only 1 Asian patient was enrolled (Table 1). Patients completed a median of 2 treatment cycles in both arms and received a maximum of 8 and 4 cycles in the QoD and QD arms, respectively.
Disease progression (53 patients, 72.6 %) was the most frequent reason for treatment discontinuation in both arms. On QoD, 31 patients (75.6 %) discontinued due to PD, 3 patients (7.3 %) due to AEs (asthenia, urinary infection, diarrhea), 2 patients (4.9 %) due to death from study disease, and 5 patients (12.2 %) due to other reasons (patient or physician decision 4, non-compliance with study procedures and treatment 1 [protocol deviation]). On QD, 22 patients (68.8 %) discontinued due to PD, 6 patients (18.8 %) due to AEs (blood bilirubin increased, colonic obstruction, worsening of ECOG performance status, QT prolongation, pyrexia, abnormal hepatic function), and 4 patients (12.5 %) due to other reasons (patient or physician decision 3, loss to follow-up 1).
DLTs and MTD
In the QoD arm, no DLTs were observed during the dose-escalation phase up to the 90 mg dose-level (Table 2). At the 100 mg dose-level, 3 patients experienced DLTs (G3 diarrhea, G3 SGOT increased; Table 2) that led to consideration of the 90 mg-dose as the MTD. Among the 12 additional patients then enrolled at this dose-level in the expansion cohort, only 1 additional DLT (G3 diarrhea) was reported, confirming the 90 mg dose as MTD for the QoD schedule.
In the QD arm, 1 of the first 3 patients treated at the 60 mg dose-level presented with a DLT of G3 SGOT increased. Four additional patients were then treated at this dose-level with no further DLTs. At the 70 mg dose-level, no DLT was observed in 3 patients. At the 90 mg dose-level, 2 of 7 patients experienced DLTs including G3 diarrhea, G3 edema limb, and G3 liver dysfunction, leading to the consideration of the 80 mg dose as MTD. One of the 3 first patients treated at the 80 mg dose-level experienced dose limiting G3 QTc prolongations. The cohort was therefore expanded to 6 more patients who had no further DLT. Further enrolment into the expanded 80 mg-cohort of the QD arm was stopped as soon as the higher MTD of 90 mg had been confirmed for the QoD arm.
General safety assessment
Serious AEs were reported in a total of 22 patients (55.7 %) in the QoD arm, and 17 patients (53.1 %) in the QD arm. Dose omissions due to AEs were required for 2 patients in the QoD arm (due to AST increased and peripheral edema) and 5 patients in the QD arm (4 due to influenza-like illness, 1 due to pneumonia). Three patients in the QoD arm required dose reductions due to AEs (2 due to diarrhea, 1 due to AST increased) but none in the QD arm.
AEs of any grade possibly related to study drug were reported by 39 patients (95.1 %) in the QoD arm, and by 29 patients (90.6 %) in the QD arm. Nausea (43.9 %), vomiting (36.6 %), pyrexia (31.7 %), asthenia (26.8 %), and decreased appetite (26.8 %) were most frequently reported in the QoD arm. Pyrexia (46.9 %), nausea (34.4 %), and asthenia, chills, diarrhea, vomiting, and AST/ALT increased (21.9 % each) were most frequently reported in the QD arm. G3/4 possibly related AEs were reported by 12 patients (29.3 %) in the QoD arm, and by 8 patients (25.0 %) in the QD arm.
Lymphocytopenia and changes in prothrombin time were the most frequent G3/4 hematologic toxicities (Table 3). Two patients in the QoD arm and 1 patient in the QD arm required packed red blood cell transfusions.
Common non-hematologic G3/4 laboratory toxicities included hepatic enzyme changes (gamma-glutamyltransferase, alkaline phosphatase, AST, ALT) and hyponatremia (Table 3).
ECG assessments revealed Fridericia-corrected QT (QTcF) intervals >450 msec for 6 patients in the QoD arm (5 at doses ≤90 mg) and for 1 patient in the QD arm (80 mg dose). Two patients in the QoD arm (both at doses ≤90 mg) and 1 in the QD arm (80 mg dose) showed mean increases of QTcF by >30 msec from baseline.
Tumor response
In the QoD arm, 7 of the 29 patients (24.1 %) enrolled during the dose escalation phase experienced clinical benefit and achieved stable disease (SD), including 3 patients with SD for at least 4 months. These patients had received doses between 50 and 70 mg of LY2334737. There was no complete response (CR) or partial response (PR), 7 patients in the QoD arm had unknown response. In the QD arm, 4 of 32 patients (12.5 %) achieved SD, including 1 patient with SD for at least 4 months. Again, there was no CR or PR, 11 patients in the QD arm had unknown response.
Pharmacokinetics
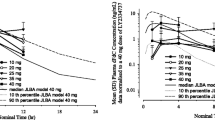
After single-dose administration of LY2334737, the areas under the curve (AUCs) of LY2334737 and its metabolite dFdC showed a linear, dose-proportional increase (Table S1 in Online Resource 2, Fig. 1a). Moderate to high apparent clearance (geometric mean ranging from 193 to 324 L/h), and a high apparent volume of distribution (geometric mean ranging from 978 to 1480 L) were observed for LY2334737. Time concentration profiles of LY2334737 and its metabolite dFdC were consistent with a time- and dose-independent clearance and distribution, leading to dose-proportional, linear increases in LY2334737 and dFdC exposure (Fig. 1a). Data were consistent with the PK information from the first-in-human Study [13] (Fig. 1b).

LY2334737 and dFdC concentration versus time profiles (means ± standard deviation) after a single dose of LY2334737 (a), dose-normalized LY2334737 and dFdC concentration in comparison to simulated profiles (median, 10th-90th percentile) from the first-in-human study PK model (b), and dFdU concentration versus time profiles after single and repeated dosing of LY2334737 in the QoD arm (c) and QD arm (d). Abbreviations: dFdC gemcitabine, 2’, 2’-difluorodeoxycytidine, dFdU difluorodeoxyuridine, hr hours, PK pharmacokinetics, QD once daily, QoD every other day, SD standard deviation
Steady state PK parameters of LY2334737 and the metabolites dFdC and dFdU are summarized in Table S2 (Online Resource 2). As anticipated from the known long half-life of dFdU, dFdU accumulated both after repeated QoD and QD dosing (Fig. 1c and d). In the QoD arm, the mean dFdU accumulation ratio, based on AUC0-24 ratios of Day 21/Day 1, was 3.04 (coefficient of variation [CV] 38.2 %; 90 % confidence interval [CI] 2.66–3.44; n = 26). The corresponding accumulation ratio for the QD arm was 4.21 (CV 31 %); 90 % CI 3.72–4.70; n = 21).
Incorporation of dFdC into DNA
Figure 2 shows the time- and dose-related increase in dFdC incorporation into patients’ DNA isolated from whole blood following treatment with LY2334737. In both arms, dFdC incorporation had decreased on Day 28 after the 1-week rest from treatment and showed a trend towards saturation during the second 28-day treatment cycle.

Amount of dFdC incorporated into DNA (extracted from whole blood), expressed as pg of dFdC per μg of deoxyguanosine (dFdC/dG). Graphs a and b show mean (SD) dFdC/dG ratios over time for the QoD (a) and QD (b) treatment arms. The grey shaded areas indicate LY2334737 treatment periods. Graph c shows the mean (SD) dFdC/dG ratios concentration on Day 21 over dose (lower graphs). Abbreviations: dFdC gemcitabine, 2’, 2’-difluorodeoxycytidine, dG deoxyguanosine, DNA deoxyribonucleic acid, QD once daily, QoD every other day, SD standard deviation
CK18 M30 and M65 antigen expression
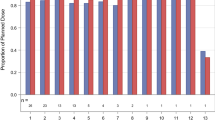
In both arms, median levels of the fragmented CK18 M30 antigen increased over time (Online Resource 3, Fig. S1a and b). Changes were less pronounced for total CK18 M65 antigen (Online Resource 3, Fig. S1c and d). Maximum increases of CK18 M30 by approximately 100 % were reached after 21 days of QoD administration (Online Resource 3, Fig. S1a), there was no clear dose response relationship. On Day 21, median CK18 M30 increases ranged from 30 % (80 mg dose) to 100 % (40 mg and 90 mg doses) in the QoD arm, and from 0 % (80 mg dose) to 60 % (40 mg dose) in the QD arm (Fig. 3).

Percentage change in median cytokeratin 18 M30 antigen levels from baseline on Day 21. Abbreviations: n number of patients per dose group, QD once daily, QoD every other day, SD standard deviation
Pharmacogenetics
Genomic DNA samples for SNP assessment were available from 45 patients treated with LY2334737. These included 41 patients (91.1 %) of Caucasian origin and no Asian patients. One SNP (rs4148323) was found to be mono-allelic in the mainly Caucasian cohort tested and therefore excluded from further analysis. Allelic distributions for the 6 remaining SNPs were consistent with the HapMap CEU cohort data, all SNPs tested were within HWE.
The homozygote AA genotype of the SNP rs818202 in the CDA gene was found to be significantly associated with Level 3 hepatotoxicity (≥G3 increase of either AST, ALT, or bilirubin) under allelic, genotypic and recessive models, both for all patients and for Caucasians only (e.g., genotypic model: all patients: corrected p = 0.006; Caucasians only: corrected p = 0.012). However, the AA-group included 4 patients only (all Caucasian). Of these 4 patients, 3 experienced Level 3 hepatotoxicity (recessive model, Caucasians only: p = 0.007), 2 of them had ≥ G3 AST increase (p = 0.041; Table S3 in Online Resource 2, 1 had bilirubin G3 toxicity). The distribution of maximum AST, ALT, and bilirubin values in the overall population also revealed higher values in the AA subgroup when compared to the AG and GG genotype groups (Online Resource 3, Fig. S2). A post-hoc ENCODE analysis revealed that the rs818202 SNP resides in an open chromatin region of the CDA gene, in a promoter and regulatory enhancer region with many transcription factor binding sites.
Additionally, there was moderate evidence for 1 of the HLA SNPs (rs3096691) to be associated with Level 2 hepatotoxicity under the allelic model only (all patients: uncorrected p = 0.028, corrected p = 0.168; Caucasians only: uncorrected p = 0.019, corrected p = 0.114). No significant associations with safety events were found for the remaining 4 SNPs assessed (CES2 – rs2303218; HLA – rs12526186; rs2269706; and SLC28A1 – rs45523532).
No clustered AEs as previously observed in Asian patients [14], i.e., thrombocytopenia and concurrent ≥ G3 increase of AST, ALT, or bilirubin, were identified in this study.
Discussion
This Phase I study established and confirmed the recommended Phase II dose and schedule for the oral gemcitabine prodrug LY2334737 at 90 mg given QoD for 21 days, followed by 7 drug-free days. A single DLT (G3 diarrhea) was reported among the 17 patients who received the recommended dose and schedule. The alternative 28-day schedule where LY2334737 was given QD every other week was less well tolerated, with 2 of 7 patients reporting 3 DLTs on the 90 mg dose-level (G3 diarrhea, G3 limb edema, G3 hepatic failure).
Use of the QoD schedule reported in this study may therefore more than double the MTD compared with the previously established MTD for LY2334737 of 40 mg QD for 14 days, followed by 7 drug-free days [13]. Previous findings from 2 other Phase I studies, where LY2334737 was either combined with docetaxel [22] or capecitabine [23], indicated that a QD regimen of LY2334737 provided suboptimal exposure and unacceptable toxicity.
At the recommended dose in both schedules of 90 mg QoD ×21 days, the main toxicities were vomiting and nausea. Hepatic dysfunction was reported, consistent with the known effects of gemcitabine on hepatic function [1]. However, no hepatotoxicity was noted, and no clustering of hepatotoxicity with thrombocytopenia, as found with lower doses of LY2334737 in Asian patients [14], was observed in this study of mainly non-Asian patients. This observation suggests that genomic differences between Asian and Caucasian patients may influence metabolism and tolerability of LY2334737, as described below.
Oral LY2334737 showed a linear PK in both schedules. The concentration-time profiles of LY2334737 and its metabolite dFdC were consistent with previously reported profiles [13] and showed time- and dose-independent clearance and distribution, leading to dose-proportional increases in LY2334737 and dFdC exposure.
The long half-life of the metabolite dFdU, and its accumulation after repeated dosing, was consistent with the observation in the first-in-human study [13]. Exposure to dFdU results from both pre-systemic-first pass liver metabolism and post-systemic metabolism of LY2334737 and dFdC. First-pass metabolism is the rate-limiting step [8]. The greater the first-pass liver metabolism of dFdC, the higher the dFdU accumulation ratio observed was, after repeated dosing: After 21 days QoD treatment with LY2334737, the mean accumulation ratio (Day 21/Day 1) in our study was 3.0, compared with a ratio of 5.4 after 21 days QoD treatment with oral gemcitabine [8].
Increased plasma levels of CK18 antigens, released from intracellular CK18 filaments upon cell death, have been associated with better clinical response [17]. CK18 has also shown a potential as prognostic marker in non-small cell lung cancer (NSCLC) patients [18]. Total CK18 (M65 antigen) has been used as a serum biomarker for carcinoma cell death, whereas the enzyme-cleaved fragment CK18 M30 antigen is a known marker of tumor cell apoptosis [18, 19]. We observed a maximum increase of the CK18 M30 antigen by approximately 100 % after 21 days of LY2334737 given QoD, indicating that LY2334737 demonstrated a pro-apoptotic effect across the dose range tested. The lack of a clear dose-response relationship may indicate that the maximum effect on the M30 marker of anti-tumor activity might already be reached at the 40 mg dose. The anti-tumor effect of LY2334737 is mediated by incorporation of dFdC triphosphate into the DNA. The activity of the cytidine kinase which phosphorylates dFdC into dFdC triphosphate is saturable, and a trend towards saturation of dFdC incorporation was reached towards the second 28-day treatment cycle. The concentration-limited saturable formation of dFdC triphosphate may also explain the lack of dose relationship for the CK18-M30 response. No correlation was possible with clinical activity as evidence of tumor regression was limited in this heavily pretreated population.
One polymorphism, the homozygote AA genotype of the rs818202 SNP of the CDA gene, was consistently associated with Level 3 hepatotoxicity across all assays. CDA is involved in the salvaging of pyrimidines, and plays a key role in detoxifying gemcitabine [9]. The CDA gene has 1118 publically known variants, of which 1015 are intronic [24]. The more commonly studied variants, CDA*2 and CDA*3, have been associated with toxicity mainly in the Japanese population [25]. The functional relevance and clinical utility of the rs818202 SNP was previously unknown. Our post-hoc ENCODE analysis located the SNP in a promoter and regulatory enhancer region of intron 2 with many transcription factor binding sites. This may indicate some functional characteristics, and potentially links the DNA change to transcriptional regulation of CDA. However, there are several limitations and caveats regarding the pharmacogenetic data obtained in this study. The 7 different SNPs assessed were selected because they were previously associated with safety events identified in Asian LY2334737-treated patients who had received LY2334737 in different treatment combinations (data on file). Also, the pharmacogenetic analyses did not adjust for the different tumor types, doses, and schedules of LY2334737 used.
In conclusion, both schedules of LY2334737 evaluated in this Phase I study displayed linear PK and acceptable safety profiles in patients with advanced metastatic solid tumors. The recommended dose and schedule of LY2334737 for subsequent Phase II studies, in Caucasian patients, would be 90 mg given QoD for 21 days, followed by 7 drug-free days. The QD schedule was less well tolerated. The potential association between the AA genotype of the rs818202 SNP and hepatic toxicity of LY2334737 found in this study may require further evaluations. The totality of the data collected across the LY2334737 program has not led to a clear differentiation when compared with intravenous gemcitabine, which is an established agent with demonstrated efficacy across a wide range of tumors. Further development of LY2334737 might include combination studies with oral agents such as capecitabine, based on pre-clinical research indicating a synergistic effect [7].”
References
Gemzar Summary of Product Characteristics (SPC) http://agence-prd.ansm.sante.fr/html/par_eu/20110907_fr390_gemcitabinemylan_spc.pdf. Accessed 24 November 2014
Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN (1991) A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9:491–498
Patel SR, Gandhi V, Jenkins J, Papadopolous N, Burgess MA, Plager C, Plunkett W, Benjamin RS (2001) Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol 19:3483–3489
Tempero M, Plunkett W, Ruiz Van Haperen V, Hainsworth J, Hochster H, Lenzi R, Abbruzzese J (2003) Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 21:3402–3408
Sakamoto H, Kitano M, Suetomi Y, Takeyama Y, Ohyanagi H, Nakai T, Yasuda C, Kudo M (2006) Comparison of standard-dose and low-dose gemcitabine regimens in pancreatic adenocarcinoma patients: a prospective randomized trial. J Gastroenterol 41:70–76
Takahashi Y, Mai M, Sawabu N, Nishioka K (2005) A pilot study of individualized maximum repeatable dose (iMRD), a new dose finding system, of weekly gemcitabine for patients with metastatic pancreas cancer. Pancreas 30:206–210
Pratt SE, Durland-Busbice S, Shepard RL, Donoho GP, Starling JJ, Wickremsinhe ER, Perkins EJ, Dantzig AH (2013) Efficacy of low-dose oral metronomic dosing of the prodrug of gemcitabine, LY2334737, in human tumor xenografts. Mol Cancer Ther 12:481–490
Veltkamp SA, Jansen RS, Callies S, Pluim D, Visseren-Grul CM, Rosing H, Kloeker-Rhoades S, Andre VA, Beijnen JH, Slapak CA, Schellens JH (2008) Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res 14:3477–3486
Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW (1992) Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab Dispos 20:849–855
Moysan E, Bastiat G, Benoit JP (2013) Gemcitabine versus modified gemcitabine: a review of several promising chemical modifications. Mol Pharm 10:430–444
Bender DM, Bao J, Dantzig AH, Diseroad WD, Law KL, Magnus NA, Peterson JA, Perkins EJ, Pu YJ, Reutzel-Edens SM, Remick DM, Starling JJ, Stephenson GA, Vaid RK, Zhang D, McCarthy JR (2009) Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J Med Chem 52:6958–6961
Pratt SE, Durland-Busbice S, Shepard RL, Heinz-Taheny K, Iversen PW, Dantzig AH (2013) Human carboxylesterase 2 hydrolyzes the prodrug of gemcitabine (LY2334737) and confers prodrug sensitivity to cancer cells. Clin Cancer Res 19:1159–1168
Koolen SL, Witteveen PO, Jansen RS, Langenberg MH, Kronemeijer RH, Nol A, Garcia-Ribas I, Callies S, Benhadji KA, Slapak CA, Beijnen JH, Voest EE, Schellens JH (2011) Phase I study of oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin Cancer Res 17:6071–6082
Yamamoto N, Nokihara H, Yamada Y, Uenaka K, Sekiguchi R, Makiuchi T, Slapak CA, Benhadji KA, Tamura T (2013) Phase I study of oral gemcitabine prodrug (LY2334737) in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 71:1645–1655
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Wickremsinhe ER, Lutzke BS, Jones BR, Schultz GA, Freeman AB, Pratt SE, Bones AM, Ackermann BL (2010) Quantification of gemcitabine incorporation into human DNA by LC/MS/MS as a surrogate measure for target engagement. Anal Chem 82:6576–6583
Ueno T, Toi M, Linder S (2005) Detection of epithelial cell death in the body by cytokeratin 18 measurement. Biomed Pharmacother 59(Suppl 2):S359–S362
De Petris L, Brandén E, Herrmann R, Sanchez BC, Koyi H, Linderholm B, Lewensohn R, Linder S, Lehtiö J (2011) Diagnostic and prognostic role of plasma levels of two forms of cytokeratin 18 in patients with non-small-cell lung cancer. Eur J Cancer 47:131–137
Linder S, Olofsson MH, Herrmann R, Ulukaya E (2010) Utilization of cytokeratin-based biomarkers for pharmacodynamic studies. Expert Rev Mol Diagn 10:353–359
Sasieni PD (1997) From genotypes to genes: doubling the sample size. Biometrics 53:1253–1261
ENCODE Project Consortium (2011) A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9:e1001046. doi:10.1371/journal.pbio.1001046
Salazar R, Morales S, Gil-Martín M, Aguirre E, Oaknin A, Garcia M, Callies S, Wickremsinhe ER, Benhadji KA, Llombart A (2014) Phase 1 dose escalation and pharmacokinetic evaluation of oral gemcitabine prodrug (LY2334737) in combination with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 73:1205–1215
Infante JR, Benhadji KA, Dy G, Fetterly G, Wee Ma W, Callies S, Adjei AA (2015) Phase 1 study of oral gemcitabine prodrug LY2334737 in combination with capecitabine in patients with advanced solid tumors. Investig New Drugs 33:432–439
Iyer SN, Ankala A, Singhal RS, Hegde MR (2013) Determination of common genetic variants in cytidine deaminase (CDA) gene in Indian ethnic population. Gene 524:35–39
Sugiyama E, Kaniwa N, Kim SR, Kikura-Hanajiri R, Hasegawa R, Maekawa K, Saito Y, Ozawa S, Sawada J, Kamatani N, Furuse J, Ishii H, Yoshida T, Ueno H, Okusaka T, Saijo N (2007) Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J Clin Oncol 25:32–42
Acknowledgments
This work was supported by Eli Lilly and Company, Indianapolis, Indiana, USA. We would like to thank all patients for participating in the study. Karin Helsberg and Annemarie Huetz, Trilogy Writing & Consulting, Frankfurt, Germany, provided medical writing support on behalf of Eli Lilly and Company.
Conflicts of interest
ER Wickremsinhe, CL Horn, H Ouyang, S Callies, and KA Benhadji are Eli Lilly and Company employees, SP Myrand has been employed by Eli Lilly and Company at the time the research has been done. ER Wickremsinhe, S Callies, and KA Benhadji also own Eli Lilly stock. SJ Faivre, AJ Olszanski, K Weigang-Köhler, H Riess, RB Cohen, CL Horn, and E Raymond have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Faivre, S.J., Olszanski, A.J., Weigang-Köhler, K. et al. Phase I dose escalation and pharmacokinetic evaluation of two different schedules of LY2334737, an oral gemcitabine prodrug, in patients with advanced solid tumors. Invest New Drugs 33, 1206–1216 (2015). https://doi.org/10.1007/s10637-015-0286-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0286-7