
Abstract
Background
Veliparib (ABT-888) is an oral PARP inhibitor expected to increase gemcitabine activity. This phase I determined the maximal tolerable dose (MTD), dose-limiting toxicities (DLT), antitumor activity, pharmacokinetics (PK), and pharmacodynamics (PD) of veliparib combined with gemcitabine.
Methods
Patients with advanced solid tumors received veliparib (10–40-mg PO BID) on chemotherapy weeks with gemcitabine 500–750-mg/m2 IV on days 1, 8, and 15 (28-day cycle), or on days 1 and 8 (21-day cycle). The MTD, DLT, adverse events, PK, and PD were evaluated.
Results
Eleven patients were enrolled on the 28-day schedule. The 28-day schedule was considered intolerable and amended to a 21-day schedule, with 20 patients enrolled. Grade ≥ 3 adverse events were myelosuppression-related. The MTD was determined to be 750-mg/m2 gemcitabine IV on days 1 and 8- and 20-mg PO veliparib BID days 1–14 on a 21-day schedule. Of 27 patients evaluable for response, 3 had PR and 15 had SD. There was no evidence of any major drug–drug interaction, and PK parameter values for veliparib, gemcitabine, and dFdU were as expected. Analysis of PBMCs showed evidence of PARP inhibition and DNA damage associated with therapy.
Conclusions
Gemcitabine at 750-mg/m2 IV on days 1 and 8 combined with veliparib at a dose of 20-mg PO BID days 1–14 on a 21-day schedule is relatively well-tolerated, with manageable, expected toxicities. Clinical responses were observed in a pretreated population of patients, suggesting that this combination should be further evaluated in the phase II setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gemcitabine is a deoxycytidine nucleoside analog that is FDA-approved for a wide range of solid tumors, including pancreatic, non-small cell lung, ovarian, and breast cancer. As a result of its clinical activity and manageable safety profile, it has been investigated in combination with new cytotoxic and/or biologic therapies for the treatment of several solid tumors [1]. Gemcitabine exhibits cell-phase specificity, primarily killing cells in S-phase and inhibiting progression through G1/S-phase. Gemcitabine is metabolized intracellularly by deoxycytidine kinase to the monophosphate (dFdCMP) metabolite and then by other kinases to the active diphosphate (dFdCDP) and triphosphate (dFdCTP) nucleotides. Gemcitabine cytotoxicity is attributed to a combination of the biological activities of the diphosphate and triphosphate metabolites, respectively, leading to inhibition of DNA synthesis. dFdCDP inhibits ribonucleotide reductase, which is responsible for catalyzing the generation of deoxynucleoside triphosphates (dNTPs) for DNA synthesis. Inhibition of this enzyme by dFdCDP reduces intracellular deoxynucleotide concentrations, including dCTP. dFdCTP competes with dCTP for incorporation into cellular DNA. Reduction of dCTP intracellular concentration, by the action of dFdCDP, enhances DNA incorporation of dFdCTP. Once dFdCTP is incorporated, one additional nucleotide is added to the DNA, after which DNA synthesis stops. DNA polymerase epsilon is unable to remove the gemcitabine nucleotide and repair the growing DNA strands (masked chain termination) [2,3,4]. There is evidence that gemcitabine causes DNA damage including double-strand breaks, which contributes to its cytotoxicity [5,6,7]. Once incorporated, gemcitabine causes topoisomerase I trapping and formation of topoisomerase I-DNA–gemcitabine cleavage complexes, which can be repaired by the action of poly(adenosine diphosphate [ADP]-ribose) (PAR) polymerase (PARP)-1 and -2 [8, 9]. Therefore, the inhibition of PARP-1 and -2 by veliparib (ABT-888) could prevent the reversal of topoisomerase I-DNA–gemcitabine complexes and enhance gemcitabine cytotoxicity. Another mechanism for repair of these topoisomerase I-DNA cleavage complexes involves tyrosyl-DNA phosphodiesterase 1 (TDP1) that removes topoisomerase I from the 3′ terminus of DNA [8]. This repair causes a single-strand break that needs to be processed by base excision repair (BER). Given the major role of PARP in BER, this represents another mechanism by which veliparib could increase gemcitabine cytotoxicity. Indeed, there is evidence that BER and more specifically, apurinic/apyrimidinic endonuclease (APE) are actively involved in repairing gemcitabine-related DNA damage. Studies have shown a 100-fold enhancement of gemcitabine cytotoxicity by an APE antisense molecule in PANC-1 cells [10]. Enhancement of gemcitabine antitumor effects by PARP inhibition with 3-aminobenzamide in pancreatic cancer models has been reported in both in vitro and in vivo murine xenograft models [11].
Elevated PARP levels in cancer cells compared to normal cells correlates with drug resistance and with an increased overall ability of cancer cells to survive genotoxic stress [12]. Veliparib is an inhibitor of PARP-1 and -2 with good oral bioavailability [13]. In mouse models of melanoma, glioma, and breast cancer, veliparib enhanced the cytotoxic effects of several chemotherapeutic agents (temozolomide, cisplatin, carboplatin, and cyclophosphamide) [14].
Taken together, these pre-clinical results formed the scientific rationale to support the hypothesis that veliparib might be able to potentiate the cytotoxic effects of gemcitabine in several malignancies, where gemcitabine is clinically active. Herein, we report an NCI-CTEP sponsored phase I trial (NCI 8324) combining gemcitabine with veliparib, with primary endpoints of identifying the maximum tolerated dose (MTD) and associated dose-limiting toxicities (DLTs). Secondary objectives were to establish the safety and tolerability of the combination, to determine the PK and PD of veliparib and gemcitabine when administered in combination, and to document preliminary clinical efficacy.
Methods
Patient selection
Study eligibility required patients with histologically documented solid tumors, progressed on standard therapy or without acceptable standard therapeutic options. Patients had to be ≥18 years, provide written informed consent, have an ECOG PS ≤2, and have a life expectancy of ≥3 months. Eligibility required adequate marrow, renal, and hepatic function, and a washout period of 4 weeks (6 weeks for mitomycin C or nitrosoureas).
Treatment plan
This was a multicenter, NCI-CTEP-sponsored trial performed at the University of Pittsburgh Cancer Institute and UPMC Hillman Cancer Center, Pittsburgh, Pennsylvania and at the Penn State Cancer Institute, Hershey, Pennsylvania, registered under ClinicalTrials.gov Identifier: NCT01154426.
Veliparib was initially administered orally 10-mg BID on days 1 through 21 of a 28-day cycle with a 1-week rest. Gemcitabine was administered as a 30-min infusion weekly for 3 weeks at 750 or 500 mg/m2 on dose levels (DL) 1 and −1, respectively, followed by a 1-week rest. For cycle 1 only, veliparib was planned for administration at the start of the gemcitabine infusion (days 1, 8, and 15). Because of significant myelosuppression associated with this 28-day schedule, we subsequently modified the schedule to a 21-day cycle with 14 days of therapy followed by a 1-week rest. Table 1 presents the dosing schema, where doses of veliparib were 20-, 20-, or 40-mg veliparib BID with gemcitabine weekly for 2 weeks at 500, 750, or 750 mg/m2 on DL 1, 2, and 3, respectively.
During cycle 1, veliparib was also dosed on days −2 and −1 to allow PK and PD studies.
After achieving the first two DLTs at one DL, all subsequent patients without a known BRCA mutation were to be screened with the BRCAPRO computer program to assess the likelihood of having a BRCA mutation [15]. Patients with a BRCAPRO likelihood >20% of having a BRCA mutation were counseled and referred for BRCA testing. Dose escalation on the 28-day schedule was to be split into two cohorts (BRCA mutant versus BRCA wild type), with each cohort having a separate dose modification schema, to determine whether or not BRCA status influenced toxicity. This was done to address the concern that BRCA-mutated patients might be at increased risk of toxicity with the combination of a PARP inhibitor and a chemotherapeutic agent.
The study was approved by the respective institutional review boards and ethics committees, and patient accrual was initiated in May 2010.
Safety assessments
Safety evaluations were conducted at baseline and weekly thereafter. Patients were evaluated with a medical history and physical examination and a laboratory panel including a complete blood count and serum chemistries, which included hepatic and renal function tests. On the ‘off’ week, laboratory blood work was repeated. Adverse events were assessed weekly and graded according to the Common Terminology Criteria for Adverse Events (version 4.0).
This phase I trial followed a standard 3 + 3 design [16]. Dose escalation was allowed when no DLT was observed in three patients or when no more than 1 DLT was observed in six patients. If a dose was held in cycle 1 because of a non-drug-related event, the patient was unevaluable for DLT and was replaced. The maximum tolerable dose (MTD) was the highest dose level at which no more than one of six patients experienced dose-limiting toxicities (DLT). The MTD was determined by drug-related DLTs occurring during cycle 1 only. DLT definition included any grade 4 or higher hematologic toxicity and grade 3 febrile neutropenia and any grade 3 or higher non-hematologic toxicity with the following clarifications: grade 3 diarrhea only when refractory to supportive care; grade 3 nausea and vomiting or rise in creatinine only if unable to correct to grade ≤ 1 within 24 h; and grade 3 metabolic toxicities only if unable to correct to grade ≤ 2 within 24 h. A delay in starting cycle 2 by more than 2 weeks due to toxicity, regardless of attribution or grade, was considered a DLT. Inability to deliver cycle 1 day 8 or 15 gemcitabine was also considered a DLT.
Once the MTD was established, this cohort was expanded to a total of 12 patients to further characterize the toxicities associated with that particular dose level and to allow additional PK and PD studies. Data were collected, entered into Theradex, and analyzed by the authors.
Tumor response assessment
Although measureable disease was not an eligibility criterion for accrual on this trial, wherever possible, tumor measurements were taken pre-treatment and repeated every two cycles. Responses were graded according to the Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 [17]. Where appropriate, disease evaluation with cancer antigen 125 (CA-125) was assessed according to Gynecologic Cancer Intergroup criteria (GCIG) [18]. Only those patients who had measurable disease present at baseline, received at least one cycle of therapy, and had their disease re-evaluated were considered evaluable for response.
PK and PD
PK and PD studies were performed during cycle 1, as described in Supplementary Methods.
Ascites case
A patient with metastatic adenocarcinoma of the pancreas and ascites enrolled on this trial and was studied for PK and PD of his ascites, as described in Supplementary Methods.
Cytidine deaminase activity
Cytidine deaminase (CDA) enzymatic activity was determined in plasma at pre-treatment and on day 1 1.5-h post-veliparib, as described in Supplementary Methods.
Statistical analysis
SAS software (version 9.4) was used to analyze demographic, adverse events, and efficacy data. Patients enrolled but who were not administered any treatment were excluded from all analyses. Patients who received any study treatment were evaluable for toxicities. Adverse events that were possibly, probably, or definitely related to treatment were considered.
For PK and PD data, Jonckheere–Terpstra and Wilcox signed rank tests were performed with IBM SPSS Statistics version 22 (Armonk, New York).
Results
Patients
Eleven patients were enrolled on the 28-day schedule and 20 patients on the 21-day schedule, with baseline characteristics, as presented in Suppl. Table 1. The median age of the patients was 57 years (range 39–73), patients had good performance status (84% ECOG ≤ 1), and predominant cancer types being lung (29%), breast (26%), and pancreas (19%). The majority of patients had received one or two lines of previous therapy.
A total of 187 cycles were administered.
DLTs and MTD
On the 28-day schedule, 10 of the 11 patients enrolled were evaluable for DLT, with one patient hospitalized during cycle 1 for an unrelated pulmonary embolism resulting in inability to continue therapy. Three patients were treated at DL1. Two of the first three patients were evaluable for DLT and experienced DLTs (grade 3 thrombocytopenia and grade 3 neutropenia; both patients were not able to receive day 15 gemcitabine) (Table 1). One of these two patients had BRCA-mutated ovarian cancer. The BRCA status of the other patient was unknown. In view of these results, as per protocol, cohorts were then split based upon BRCA status. Dosage was decreased to DL-1. At this dose level, one of the first three BRCAPRO negative patients experienced a DLT (grade 3 neutropenia with inability to deliver day 8 gemcitabine). This cohort was then expanded as per study protocol. Of the next four BRCAPRO negative evaluable patients, there was an additional DLT (grade 3 nausea and vomiting) suggesting that DL-1 was not clinically tolerable in BRCA-negative patients. One BRCA-mutated patient was enrolled at DL-1, and experienced a DLT (grade 3 neutropenia with inability to deliver day 15 gemcitabine). A reduction in the gemcitabine dose to less than 500 mg/m2 was considered to result in sub-therapeutic drug exposures, and therefore, instead of accruing additional patients, we elected to modify the study protocol.
In the hopes of developing a treatment protocol that was (a) deliverable, (b) that administered doses of gemcitabine thought to be within the therapeutic range, and (c) that allowed for the investigation of the combination of gemcitabine and veliparib at multiple dose levels, the protocol was subsequently amended. Patients with more than two prior chemotherapeutic regimens were excluded due to the significant hematologic toxicity observed with the 28-day schedule, and we modified the treatment regimen to a 21-day schedule (gemcitabine days 1 and 8, veliparib BID × 14 days). BRCAPRO screening and BRCA testing were still required, although cohorts were no longer split based upon BRCA status.
On the 21-day schedule, 18 of the 20 patients enrolled were evaluable for DLT, with one patient withdrawing consent at the end of cycle 1, and one patient withdrawing for surgical intervention. Two patients were enrolled with more than two previous lines of therapy, but they did not experience DLT in cycle 1. Three patients tolerated treatment at DL1 without a DLT prompting dose escalation. Three patients were treated at DL2 without DLT. Two of three patients treated at DL3 experienced a DLT (grade 3 neutropenia and grade 2 neutropenia; both with inability to deliver day 8 gemcitabine). Three additional patients were treated at DL2 without DLT. Therefore, DL2 declared the MTD, and enrollment to this dose level was then expanded. A total of 12 evaluable patients were eventually treated at DL2. There were two DLTs (both grade 3 neutropenia with inability to deliver day 8 gemcitabine) in this cohort, thereby confirming this dose level as the MTD. Of the patients treated at the MTD, 11 of 12 received at least 2 cycles with one patient going on to receive 14 cycles of treatment. Of the 18 evaluable patients, 3 were BRCA mutated (1 at each dose level). At DL1 and DL2, the BRCA-mutated patients (fallopian tube cancer and breast cancer, respectively) did not experience a DLT. At DL3, the BRCA-mutated patient did experience a DLT, as did a BRCAPRO negative patient.
Adverse event profile
Table 2 lists severe adverse events (grade 3/4) and Suppl. Table 2 lists common (>5%) adverse events of all grades incurred in all administered cycles. Of the 31 patients, 23 experienced grade 3/4 toxicities, of which 20 experienced grade 3/4 toxicities related to myelosuppression. In the 28-day patient cohort (11 patients), gemcitabine (500 mg/m2 days 1, 8, and 15) in combination with veliparib (10-mg BID days 1–21) was not clinically tolerable with DLTs noted in 2 of 7 BRCA non-mutated and 1 of 1 BRCA-mutated patients.
Among the 18 evaluable patients on the 21-day schedule, 4 DLTs (only 1 BRCA mutated at DL3, Table 1) were observed. Two of the three evaluable patients at DL3 experienced a DLT, making this particular dose level undeliverable. DL2 (gemcitabine 750-mg/m2 days 1 and 8 and veliparib 20-mg BID days 1–14) was then determined to be the MTD, with 2 DLTs in an expanded cohort of 12 patients.
Of a total of nine patients who experienced a DLT, all but one was related to myelosuppression. There were six cases of neutropenia, one of leukopenia, and one of thrombocytopenia (Table 1). Of note, gastrointestinal (GI) toxicity was relatively mild with only one GI-related DLT (nausea and vomiting).
Antitumor activity
Of the 11 patients treated on the 28-day schedule, ten were evaluable for response. There was one partial response in a patient with pancreatic cancer and five patients with stable disease (breast cancer, 1; lung cancer, 2; ovarian cancer, 1; and pancreatic cancer, 1). Of the 20 patients treated on the 21-day schedule, 17 were evaluable for response. There were 2 partial responses (metastatic adrenocortical carcinoma, 1; ovarian carcinoma, 1) and 10 patients with stable disease (breast cancer, 3; lung cancer, 4; pancreatic cancer, 2; unknown primary, 1), see Suppl. Table 3.
Among nine lung cancer patients, six had stable disease (4–20 cycles). Of the four evaluable patients with pancreatic cancer, three had stable disease (2–6 cycles) and one had a PR (12 cycles). Four of seven patients with breast cancer had stable disease (4–14 cycles).
Of the three patients with a PR, only the ovarian cancer patient was BRCA positive (22 cycles). The other two (pancreatic cancer and adrenocortical carcinoma) were BRCA negative. The pancreatic cancer patient was treated on the 28-day schedule at DL-1. The other two patients were treated on the 21-day schedule—one at one DL below the MTD (adrenocortical carcinoma) and the other at one DL above the DLT (ovarian cancer).
Pharmacokinetics
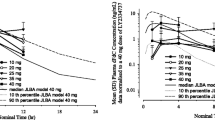
Veliparib PK data were available for 30 patients (Table 3). Data are presented by veliparib dose, combining 500- and 750-mg/m2 doses of gemcitabine, as any differential effect on veliparib PK was assumed to be minimal. Veliparib exposure increased with dose with a half-life of 5 h and an apparent clearance of 20 L/h. There was a statistically significant effect of gemcitabine on veliparib exposure, as judged by the observed accumulation index for C max and AUC0–12 relative to the theoretical accumulation index, of about 20% above the accumulation expected based on each patient’s veliparib alone half-life.
Gemcitabine PK data were available for 27 patients (Table 4). Data are presented by gemcitabine and veliparib dose. Gemcitabine exposure increased with dose. The half-life of dFdU was imprecisely estimated because of the relatively short sampling period (48 h relative to the estimated value of approximately 50 h). There appeared to be a trend towards an increased gemcitabine half-life, and lower dFdU/dFdC C max and AUC ratios with increasing veliparib dose. However, with 10, 20, and 40-mg BID veliparib, only the gemcitabine half-life increased statistically significant (23, 24, and 54 min, respectively) when compared at 750-mg/m2 gemcitabine, using the Jonckheere–Terpstra test (P = 0.034).
Pharmacodynamics
PBMC PAR levels were evaluated in five patients. Two patients were analyzed with NCI provided reagents prior to suspension of this assay by the NCI. PAR levels were quantifiable in all PBMC samples. As shown in Fig. 1, PAR levels rapidly decreased after veliparib administration in pt#1-1-13 and increased after gemcitabine infusion. PBMCs from pt#1-1-15 demonstrated no reduction in PAR levels after veliparib administration but revealed a rapid decline after gemcitabine administration. Three additional patients were evaluated for PAR levels using a commercially available ELISA kit. Of note, the basal PAR expression in these three patients was significantly lower due to the differences in the PAR standards provided in each kit used to quantitate PAR (31 versus 231, pg/107 PBMCs, respectively). In these three patients, PAR levels decreased 1 h after veliparib administration. One patient (pt#1-2-31) had an unexplainable spike in PAR levels at 6-h post-veliparib. Gemcitabine administration seemed to reduce PAR levels even further.

PAR level in PBMCs from patients treated with gemcitabine plus veliparib. Blood samples were drawn at the indicated times. PBMCs were isolated and processed for PAR ELISA. Clinical events (PR partial response, SD stable disease, DLT dose-limiting toxicity) are listed behind patient identifier
The combination of gemcitabine and veliparib is believed to enhance DNA damage. To evaluate the extent of this damage, we measured activation by phosphorylation of H2AX protein on serine 139 in PBMCs. Suppl. Figure 1 shows H2AX activation over a 24-h period post-veliparib/gemcitabine administration. H2AX was activated in eight out of ten patients, although the kinetics of the activation varied. These patients had either SD, PR, or DLT, while the two non-responding patients did not show activation of H2AX in their PBMCs. Neither the peak plasma concentration of gemcitabine nor gemcitabine AUC correlated with the level of H2AX activation after 1 h (Suppl. Figure 2). Our PD studies showed ATM activation in only three patients.
Ascites case
The concentration versus time courses of gemcitabine and dFdU in plasma and ascites are presented in Suppl. Figure 3A. The concentration versus time courses of veliparib in plasma and ascites are shown in Fig. 2, and the PK parameters are shown in Suppl. Table 4. Peak concentrations of gemcitabine in plasma and ascitic fluid were 11.8 and 1.58 µg/mL, respectively. Ascites gemcitabine concentrations declined less rapidly than those in plasma; the corresponding half-lives being 1.3 and 3.0 h. The ascites-to-plasma ratio of gemcitabine AUC was 0.67, while that of the dFdU metabolite was 0.38. Peak concentrations of veliparib in plasma and ascites (on day 1) were 87 and 44 ng/mL, respectively. The half-life of veliparib was 7.4 h in plasma and 2.6 h in ascites. The ratio of ascites veliparib AUC to plasma veliparib AUC was 0.62. It is noteworthy that the trough concentration of veliparib in ascites was higher than that in plasma. The unbound fraction of veliparib was 0.67–0.73 in plasma and 0.76–0.86 in ascites (Suppl. Table 5).

a Plasma [day −2 (multiple symbol) and day 1 (filled square)] and ascites [day 1 (filled circle)] concentrations of veliparib in the patient with ascites. b Day 1 plasma concentrations of veliparib in the patient with ascites (filled square) compared to concentrations in five patients without ascites treated at the veliparib dose level. c PARP activity in PBMCs (open square) and ascites cells (filled square) in a patient treated with 10-mg veliparib BID and 750-mg/m2 gemcitabine. Time 0 is start of gemcitabine infusion
The PAR activity in ascites cells was higher than that in PBMCs (Fig. 2c). In both PBMCs and ascites cells, PAR was increased 1 h after the start of the gemcitabine infusion, and by 6 h, the PAR levels had declined, but had not returned to baseline.
CDA activity
Veliparib, at a timepoint close to T max, had a small but statistically significant effect, causing a decrease in CDA activity (mean 7%, median 15%, P = 0.042).
No obvious correlation of CDA with gemcitabine clearance or dFdU-to-gemcitabine ratio was observed (data not shown).
Discussion
Toxicity
The 28-day schedule, DL-1, was found to be not tolerable and associated with DLT. Further dose reduction of either veliparib or gemcitabine was not considered feasible, and a new 21-day schedule was then developed. With the modified 21-day schedule, DL2 (gemcitabine 750 mg/m2 and veliparib 20-mg BID × 14 days) was found to be the MTD with only 2 of a total of 12 evaluable patients experiencing a DLT (both grade 3 neutropenia, with inability to deliver day 8 gemcitabine). Escalation to DL3 was clearly not tolerated with two of three patients experiencing a DLT. Of the 12 patients treated at the MTD, 11 received at least two cycles with one patient going on to receive 14 cycles of treatment. This finding suggested that the MTD would also be the dose recommended for phase II studies. Although the 20-mg BID dose of veliparib is low relative to the single-agent MTD of veliparib at 400-mg BID [19], this dose is clearly associated with pharmacodynamic target engagement, as shown by our PAR data, and as previously reported in the phase 0 study of veliparib [20].
In this phase I study, the toxicity profile of veliparib in combination with gemcitabine is consistent with what is typically observed with gemcitabine monotherapy, with myelosuppression (leukopenia, granulocytopenia, thrombocytopenia, and anemia) and nausea and vomiting being the most frequent adverse events [21, 22]. Liver enzyme elevation, commonly reported with gemcitabine therapy, was also observed.
There have been several studies combining a PARP inhibitor with gemcitabine, confirming enhanced toxicity. The phase I of veliparib with gemcitabine and carboplatin on a 21-day schedule resulted in an MTD of veliparib 315-mg BID, gemcitabine 800 mg/m2 on days 1 and 8, and carboplatin AUC 4 on day 1, although an upfront cycle of gemcitabine carboplatin without veliparib may have remove individuals susceptible to toxicity, and a full report is not yet published [23]. In the phase I trial of gemcitabine and olaparib, the MTD was olaparib 100-mg BID (days 1–14, per 28-day cycle) plus gemcitabine 600 mg/m2 weekly. Continuous dosing of olaparib or combination with gemcitabine at doses >600 mg/m2 was not tolerated, with hematologic toxicities being most common [24]. The phase I study of olaparib with gemcitabine and cisplatin required dose reductions and changes in schedule ultimately leading to an MTD of olaparib 100 mg once on day 1, gemcitabine 500 mg/m2 on days 1 and 8, and cisplatin 60 mg/m2 on day 1 in patients who had received no more than 2 prior severely myelosuppressive chemotherapy regimens. The combination resulted in myelosuppression even at low doses and olaparib dosed for 4 days was not tolerable [25]. Finally, a recent report describes the combination of CEP-9722, a prodrug of the PARP-1/2 inhibitor CEP-8983, combined with cisplatin and gemcitabine. The study was halted before formal establishment of the MTD. However, the severity of toxicity associated with gemcitabine and cisplatin in the lead-in cycle (without CEP-9722), particularly myelosuppression, limited the ability to administer the combination in cycle 2 in nearly half of the enrolled patients, likely resulting in a selection bias, making the tolerability of the combination with CEP-9722 difficult to assess [26].
Response
Of the 27 patients evaluable for response, three patients had a documented PR (1 pancreatic, 1 metastatic adrenocortical carcinoma, and 1 ovarian carcinoma) and 15 patients were found to have SD. Of the three patients with a PR, only the ovarian cancer patient was BRCA positive (22 cycles). The other two patients (pancreatic cancer and adrenocortical carcinoma) were BRCAPRO negative. No definitive conclusions about response and BRCA status or response and primary site of disease can be drawn from this limited data set. In a patient population that was allowed prior chemotherapy including gemcitabine, our findings suggest that the gemcitabine plus veliparib combination provides a reasonable level of clinical benefit.
PK
Veliparib PK parameters calculated in the current study [Cl/F 20.3 (8.6) L/h, t½ 5.1 (2.4) h] are similar to those previously reported in the literature (Cl/F 20.9 L/h, t½ 6.1 h) [27]. The observed accumulation index was slightly higher (approximately 20%) than expected based on the half-lives, suggesting a possible minor effect of gemcitabine on veliparib exposure; however, this was not deemed to be clinically relevant. A similar effect was observed in the phase I study of olaparib with gemcitabine and cisplatin, where a statistically significant 44.8% increase in olaparib C max was observed in combination with gemcitabine relative to monotherapy [25].
Gemcitabine clearance and C max dFdU/dFdC ratio in the current study [105 (34) L/h/m2, and 1.9 (0.8)] was similar to what has been previously reported (Cl 88 L/h/m2, C max ratio 1.6) [28]. The gemcitabine half-life is quite variable, and also dependent on the lower limit of quantitation of the assay utilized. Our assay is sensitive to methods in earlier reports, and in our data, we were able to occasionally observe the start of an additional, slow compartment. The increased gemcitabine half-life with increasing veliparib dose is mainly driven by the three patients treated at the highest dose level. There was a small but statistically significant negative effect of veliparib on CDA activity. Because of the allosteric regulation of many enzymes involved in pyrimidine metabolism by their substrates and products, it would not be surprising if veliparib, an adenosine analog, affects the intracellular fate of gemcitabine [13, 29]. Exposure to gemcitabine, as expressed by AUC, was quite consistent as shown by the similar clearance values across cohorts and in line with previous reports. It remains unclear as to whether higher doses of veliparib might result in clinically significant levels of CDA inhibition.
PD
In the relatively small PD data set, we observed evidence of target engagement of PARP by veliparib in PBMCs. We have previously shown that pATM is a marker of DNA double-strand breaks [30,31,32]. Our PD studies showed ATM activation in only three patients suggesting that this drug combination did not significantly induce double-strand breaks in PBMCs, at least over the timeframe that was studied. H2AX activation appeared to be a more robust marker than ATM activation for assessment of the level of DNA damage induced by this combination.
Ascites
Ascites is a common clinical condition in various malignancies, resulting in prolonged systemic exposures and excessive toxicities, e.g., with methotrexate [33]. A case report described gemcitabine PK in ascites after a fixed dose rate of 10 mg/m2/min [34]. To our knowledge, we present the first ascites data after the labeled 30-min administration of gemcitabine [1] and veliparib. Despite the relatively low dose of gemcitabine, its concentration in ascitic fluid approached the level at which intracellular formation of the active metabolite dFdCTP is saturated (1.5–4.5 μg/mL) [35]. Ascites did not serve as a reservoir for gemcitabine, as was reported previously [34]. However, the half-life of gemcitabine in ascites was higher than that in plasma, providing concentrations close to the levels at which intracellular formation of dFdCTP is saturated for longer. The clearance of gemcitabine agrees with values for patients without ascites, and the presence of ascites does not necessitate dose reduction.
The AUC of veliparib in plasma agrees with that observed in patients without ascites. Despite the fact that the trough concentration of veliparib in ascites was higher than that in plasma, the achieved C max in ascites (44 ng/mL) was half the C max in plasma and lower than the 51 ng/mL concentration associated with a significant reduction in tumor PAR levels in single-dose studies in mice [20]. This finding could be attributed to the relatively low dose of veliparib administered to the patient, since veliparib has been shown to inhibit PARP at the 20-mg bid dose [20]. The low concentrations of veliparib in ascites could explain the observed higher PARP activity in ascites cells compared to PBMCs. The lower AUC of veliparib in ascites relative to plasma could partly be explained by the higher protein binding in plasma relative to ascites. Unfortunately, based on our data, it is difficult to conclude if ascites serves as a depot for veliparib, since PK sampling of ascitic fluid was performed only on day 1, and did not continue after discontinuation of drug.
Conclusion
In conclusion, the combination of gemcitabine at 750-mg/m2 IV on days 1 and 8 and veliparib at a dose of 20-mg PO BID days 1–14 on a 21-day schedule is safe with manageable and expected toxicities. This combination is also associated with clinical activity, and is worthy of further evaluation in the phase II setting. A randomized phase II study of gemcitabine, cisplatin, with or without veliparib in patients with advanced pancreatic cancer and a known BRCA/PALB2 mutation is currently ongoing (ClinicalTrials.gov Identifier: NCT01585805).
References
Toschi L, Finocchiaro G, Bartolini S, Gioia V, Cappuzzo F (2005) Role of gemcitabine in cancer therapy. Future Oncol 1(1):7–17. doi:10.1517/14796694.1.1.7
Plunkett W, Huang P, Searcy CE, Gandhi V (1996) Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol 23(5 Suppl 10):3–15
Plunkett W, Huang P, Gandhi V (1995) Preclinical characteristics of gemcitabine. Anticancer Drugs 6(6):7–13
Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V (1995) Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 22(4 Suppl 11):3–10
Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW, Yu S, Pon RT, Gmeiner WH, Pommier Y (2002) Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res 8(8):2499–2504
Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 278(22):20303–20312. doi:10.1074/jbc.M300198200
Ewald B, Sampath D, Plunkett W (2007) H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther 6(4):1239–1248. doi:10.1158/1535-7163.MCT-06-0633
Malanga M, Althaus FR (2004) Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J Biol Chem 279(7):5244–5248. doi:10.1074/jbc.C300437200
Malanga M, Althaus FR (2005) The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol 83(3):354–364. doi:10.1139/o05-038
Lau JP, Weatherdon KL, Skalski V, Hedley DW (2004) Effects of gemcitabine on APE/ref-1 endonuclease activity in pancreatic cancer cells, and the therapeutic potential of antisense oligonucleotides. Br J Cancer 91(6):1166–1173. doi:10.1038/sj.bjc.6602080
Jacob DA, Bahra M, Langrehr JM, Boas-Knoop S, Stefaniak R, Davis J, Schumacher G, Lippert S, Neumann UP (2007) Combination therapy of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide and gemcitabine shows strong antitumor activity in pancreatic cancer cells. J Gastroenterol Hepatol 22(5):738–748. doi:10.1111/j.1440-1746.2006.04496.x
Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, Talbot M, Robin A, Girard P, Orear C, Lissa D, Sukkurwala AQ, Garcia P, Behnam-Motlagh P, Kohno K, Wu GS, Brenner C, Dessen P, Saparbaev M, Soria JC, Castedo M, Kroemer G (2013) Cisplatin resistance associated with PARP hyperactivation. Can Res 73(7):2271–2280. doi:10.1158/0008-5472.CAN-12-3000
Davar D, Beumer JH, Hamieh L, Tawbi H (2012) Role of PARP inhibitors in cancer biology and therapy. Curr Med Chem 19(23):3907–3921
Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, Frost DJ (2007) ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 13(9):2728–2737. doi:10.1158/1078-0432.CCR-06-3039
Euhus DM, Smith KC, Robinson L, Stucky A, Olopade OI, Cummings S, Garber JE, Chittenden A, Mills GB, Rieger P, Esserman L, Crawford B, Hughes KS, Roche CA, Ganz PA, Seldon J, Fabian CJ, Klemp J, Tomlinson G (2002) Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 94(11):844–851
Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45(3):925–937
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, Van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Rustin GJ (2003) Use of CA-125 to assess response to new agents in ovarian cancer trials. J Clin Oncol 21(10 Suppl):187s–193s. doi:10.1200/JCO.2003.01.223
Puhalla S, Beumer JH, Pahuja S, Appleman LJ, Tawbi H, Stoller R, Lee JJ, Lin Y, Kiesel B, Yu J, Tan AR, Belani CP, Chew HK, Garcia AA, Morgan R, Giranda V, Shepherd SP, Chen AP, Chu E (2014) Final results of a phase 1 study of Chronically-Dosed, single-agent veliparib (ABT-888) in patients with either BRCA 1/2–mutated cancer (BRCA+), platinum-refractory ovarian or basal-like breast cancer (BRCA-wt). In: Annual Meeting American Society of Clinical Oncology, Chiacgo, Proceedings of the American Society of Clinical Oncology (Supplement to Journal of Clinical Oncology), p. 5s
Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, Ji J, Monks A, Low JA, Chen A, Murgo AJ, Collins J, Steinberg SM, Eliopoulos H, Giranda VL, Gordon G, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH (2009) Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol 27(16):2705–2711. doi:10.1200/JCO.2008.19.7681
Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB 3rd (2002) Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: eastern Cooperative Oncology Group Trial E2297. J Clin Oncol 20(15):3270–3275
Villaruz LC, Jones H, Dacic S, Abberbock S, Kurland BF, Stabile LP, Siegfried JM, Conrads TP, Smith NR, O’Connor MJ, Pierce AJ, Bakkenist CJ (2016) ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget 7(36):57714–57725. doi:10.18632/oncotarget.9757
Bell-McGuinn KM, Gray HJ, Fleming GF, Cristea MC, Medina DM, Xiong H, Dudley MW, Dunbar M, Giranda VL, Luo Y, McKee MD, Martin LP (2013) Phase I study of ABT-888 in combination with carboplatin and gemcitabine in subjects with advanced solid tumors. In: Annual Meeting American Society of Clinical Oncology, Chicago. 15S:161S
Bendell J, O’Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, Burke W, Burris H 3rd (2015) Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol 26(4):804–811. doi:10.1093/annonc/mdu581
Khan SA, Pruitt SL, Xuan L, Gerber DE (2016) Prevalence of autoimmune disease among patients with lung cancer: implications for immunotherapy treatment options. JAMA oncology. doi:10.1001/jamaoncol.2016.2238
Bahar FG, Ohura K, Ogihara T, Imai T (2012) Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci 101(10):3979–3988. doi:10.1002/jps.23258
Salem AH, Giranda VL, Mostafa NM (2014) Population pharmacokinetic modeling of veliparib (ABT-888) in patients with non-hematologic malignancies. Clin Pharmacokinet 53(5):479–488. doi:10.1007/s40262-013-0130-1
Felici A, Di Segni S, Milella M, Colantonio S, Sperduti I, Nuvoli B, Contestabile M, Sacconi A, Zaratti M, Citro G, Cognetti F (2009) Pharmacokinetics of gemcitabine at fixed-dose rate infusion in patients with normal and impaired hepatic function. Clin Pharmacokinet 48(2):131–141. doi:10.2165/0003088-200948020-00005
Beumer JH, Eiseman JL, Parise RA, Joseph E, Covey JM, Egorin MJ (2008) Modulation of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3,4,5,6-tetrahydrouridine. Clin Cancer Res 14(11):3529–3535. doi:10.1158/1078-0432.CCR-07-4885
Bakkenist CJ, Czambel RK, Hershberger PA, Tawbi H, Beumer JH, Schmitz JC (2015) A quasi-quantitative dual multiplexed immunoblot method to simultaneously analyze ATM and H2AX Phosphorylation in human peripheral blood mononuclear cells. Oncoscience 2(5):542–554
Bakkenist CJ, Beumer JH, Schmitz JC (2015) ATM serine-1981 phosphorylation is a plausible biomarker. Cell Cycle. doi:10.1080/15384101.2015.1084205
Bakkenist CJ, Czambel RK, Clump DA, Greenberger JS, Beumer JH, Schmitz JC (2013) Radiation therapy induces the DNA damage response in peripheral blood. Oncotarget 4(8):1143–1148. doi:10.18632/oncotarget.1084
Huynh TN, Luo S, Pensinger D, Sauer JD, Tong L, Woodward JJ (2015) An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc Natl Acad Sci USA 112(7):E747–E756. doi:10.1073/pnas.1416485112
Gray EE, Treuting PM, Woodward JJ, Stetson DB (2015) Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J Immunol 195(5):1939–1943. doi:10.4049/jimmunol.1500969
Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN et al (1991) A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9(3):491–498
Parise RA, Shawaqfeh M, Egorin MJ, Beumer JH (2008) Liquid chromatography-mass spectrometric assay for the quantitation in human plasma of ABT-888, an orally available, small molecule inhibitor of poly(ADP-ribose) polymerase. J Chromatogr B 872(1–2):141–147. doi:10.1016/j.jchromb.2008.07.032
Kozo D, Ross MW, Jarrah J, Barrett M, Harney RL, Courtney JB, Baburina I, Holleran JL, Beumer JH, Peters GJ, Honeywell RJ, Salamone SJ (2017) Rapid homogeneous immunoassay to quantify gemcitabine in plasma for therapeutic drug monitoring. Ther Drug Monit 39(3):235–242. doi:10.1097/FTD.0000000000000402
Waters NJ, Jones R, Williams G, Sohal B (2008) Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci 97(10):4586–4595. doi:10.1002/jps.21317
Ciccolini J, Dahan L, Andre N, Evrard A, Duluc M, Blesius A, Yang C, Giacometti S, Brunet C, Raynal C, Ortiz A, Frances N, Iliadis A, Duffaud F, Seitz JF, Mercier C (2010) Cytidine deaminase residual activity in serum is a predictive marker of early severe toxicities in adults after gemcitabine-based chemotherapies. J Clin Oncol 28(1):160–165. doi:10.1200/JCO.2009.24.4491
Acknowledgements
The authors would like to thank all of the participating patients and their families, as well as the network of investigators, research nurses, study coordinators, and operation staffs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported in part by NCI grants U01CA099168 and UM1CA186690. This project used the UPCI Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF) and was supported in part by award P30CA047904. The project described was supported by the National Institutes of Health through Grant Number UL1TR001857. EK was supported by a scholarship from the Hellenic Society of Medical Oncology.
Conflict of interest
Jan Beumer received research support from AbbVie. Shannon Puhalla has received research support from AbbVie, Pfizer, Lilly, Novartis, Incyte, Covance-Bayer, AstraZeneca, Genentech, Medivation and has been a consultant for AbbVie, MedImmune, Celldex, Puma, Pfizer, AstraZeneca, Esai, nanostring.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This trial was registered under ClinicalTrials.gov Identifier: NCT01154426.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Stoller, R., Schmitz, J.C., Ding, F. et al. Phase I study of veliparib in combination with gemcitabine. Cancer Chemother Pharmacol 80, 631–643 (2017). https://doi.org/10.1007/s00280-017-3409-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3409-3