
Summary
Background This Phase 1b study aimed to determine the recommended Phase 2 dose of LY2334737, an oral pro-drug of gemcitabine, in combination with capecitabine, an oral pro-drug of 5-fluorouracil, in patients with advanced solid tumors. In addition, pharmacokinetics (PK) and tumor response were evaluated. Patients and methods Patients with advanced/metastatic solid tumors received 650 mg/m2 capecitabine twice daily (BID) and escalating doses of LY2334737 once daily (QD; initial dose 10 mg/day), both for 14 days followed by 7-day drug holiday. Cycles were repeated until progressive disease (PD) or unacceptable toxicity. Results Fifteen patients received a median of 2 (range 1–7) treatment cycles; 14 patients discontinued due to PD, 1 due to toxicity (pyrexia). LY2334737 doses up to 40 mg/day were explored. Three dose-limiting toxicities were reported by 2 patients (fatigue, diarrhea, hyponatremia; all Grade 3). Seven patients achieved stable disease. Enrollment was stopped after unexpected hepatic toxicities were observed with LY2334737 QD in a study of Japanese patients. PK parameters for LY2334737 were consistent with the first-in-human study of LY2334737; PK data after 14 day combination treatment revealed no drug-drug interactions between LY2334737 and capecitabine. Conclusions No drug interactions or unexpected toxicities were observed in US patients when LY2334737 at doses up to 40 mg/day was administered QD in combination with capecitabine BID; the maximum tolerated dose was not reached.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
LY2334737 is an oral pro-drug of the nucleoside analog gemcitabine (2′,2′-difluorodeoxcytidine [dFdC]) [1]. Gemcitabine is approved for intravenous treatment of various solid tumors, including pancreatic, non-small-cell lung, ovarian, bladder, and breast cancer [2]. For the pro-drug LY2334737, the active metabolite dFdC has been covalently linked to valproic acid. When LY2334737 is orally administered and absorbed in the intestine, this linkage is hydrolyzed by the enzyme carboxylesterase 2, releasing the active metabolite dFdC and valproic acid into the systemic circulation [1]. In addition to being more convenient for patients, a once daily (QD) administration of LY2334737 would enable prolonged exposure to lower doses of dFdC, expected to be more effective and less toxic than the standard once-weekly intravenous administration of gemcitabine [3]. A first-in-human Phase 1 study of LY2334737 was conducted in 65 European patients with advanced solid tumors. The maximum tolerated dose (MTD) of LY2334737 was identified as 40 mg/day when given QD for 14 days followed by a 7-day drug holiday alone or with erlotinib [4].
A synergistic anticancer effect might be achieved by combining LY2334737 with capecitabine, an oral pro-drug of 5-fluorouracil (5-FU) which is approved for gastrointestinal and breast cancer treatment [5]. In a meta-analysis of 3 randomized Phase 3 studies in patients with advanced pancreatic cancer, the combination of gemcitabine and capecitabine was associated with improved survival when compared to gemcitabine alone [6]. In pre-clinical colon cancer xenograft studies, the anti-tumor activity of LY2334737 plus an MTD of capecitabine was significantly greater than either monotherapy [3].
This Phase 1b study in patients with advanced solid tumors was designed to determine the recommended dose of LY2334737 QD in combination with capecitabine twice daily (BID) for future Phase 2 studies. In addition, the pharmacokinetic (PK) profiles of LY2334737, capecitabine, and their active metabolites dFdC and 5-FU were investigated to evaluate potential drug interactions. Pharmacodynamic parameters and tumor response were additionally assessed.
Patients and methods
This open-label, Phase 1b study of LY2334737 was conducted in combination with capecitabine, in patients with advanced solid tumors. Patients who provided written informed consent were recruited at 2 sites in the US (Nashville, TN, and Buffalo, NY) between January 2008 and May 2009. The study was performed according to the Declaration of Helsinki and approved by the responsible ethical review boards.
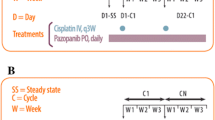
Eligible patients received oral LY2334737 (Eli Lilly and Company) QD and capecitabine (Xeloda®, Roche Pharmaceuticals) BID for 14 days, followed by a 7-day drug-free period. At the start of the first cycle, capecitabine and LY2334737 were given 24 h apart on Day 1 and Day 2, respectively, to enable investigation of monotherapy PK parameters. From Day 3 of Cycle 1 onwards, both drugs were taken on the same days. The 21-day treatment cycles were repeated until progressive disease (PD) or unacceptable toxicity.
A conventional 3 + 3 design was used for dose escalation; there was no intra-patient dose escalation [7]. The 3 initial patients received capecitabine 650 mg/m2 BID and LY2334737 10 mg QD. The LY2334737 dose was then escalated in subsequent cohorts, based on safety data and any available PK data from previous cohorts. Two additional dose escalation phases of LY2334737 were planned for higher capecitabine doses. Patients were monitored for safety on a weekly basis. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 11.0 and graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Standard laboratory tests were performed to monitor safety. Dose-limiting toxicity (DLT) was defined as occurrence of any of the following events during Cycle 1: Any non-hematological toxicity ≥ Grade (G)3 other than nausea/vomiting, G4 neutropenia lasting >4 days, >14 days needed to recover from toxicity after the last dose of Cycle 1, G3 thrombocytopenia with ≥ G2 bleeding or G4 thrombocytopenia, or any other significant drug-related toxicity considered to be dose-limiting by the investigator.
No formal efficacy analysis was performed, but lesion and response data were reported following Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 criteria [8].
Plasma concentration profiles were assessed after the initial single dose administration of each agent (monotherapy, first dose of Cycle 1), after a single dose of both agents combined (first dose of Cycle 2), and after 2 weeks of combined treatment (steady state, Day 15 of Cycle 1). Assessment of LY2334737 and dFdC plasma concentrations were performed at the following time points: At 1, 2, 4, and 8 h after administration of LY2334737 on Day 2 of Cycle 1; at 1, 2, 4, and 8 h after administration of both agents on Day 1 of Cycle 2; at pre-dose, 0.5, 1, 2, 4, 8, 24 (±2), and 48–72 h after administration of both agents on Day 15 of Cycle 1. Capecitabine and 5-FU plasma concentrations were assessed at 1, 2, and 4 h after administration of capecitabine on Day 1 of Cycle 1; at 1, 2, 4, and 8 h after administration of both agents on Day 1 of Cycle 2; at pre-dose, 0.5, 1, 2, 4, and 8 h after administration of both agents on Day 15 of Cycle 1. Plasma concentrations of LY2334737 and dFdC were assayed at Taylor Technology Inc. (now called PharmaNet USA, Inc), NJ, USA, using a validated high-pressure liquid chromatography-mass spectrometry/mass spectrometry method [9]. Capecitabine plasma concentrations were assessed by Advion BioSciences, Inc. Ithaca, NY, USA. Plasma PK parameters were analyzed by standard non-compartmental methods using WinNonlin Enterprise version 5.2. Potential drug interactions were explored by calculating the ratio between the areas under the curve (AUC) between capecitabine or LY2334737 after single-dose administration as monotherapy (on Days 1 and 2 of Cycle 1, respectively) and the respective AUC after single-dose administration of both agents combined (on Day 1 of Cycle 2).
Blood for pharmacodynamic analyses was collected on Day 1 pre-dose (baseline), and at 2 h after the Day 15 dose of Cycle 1. The incorporation of dFdC into DNA extracted from whole blood was assessed based on the ratio of dFdC to deoxyguanosine (dG) in the DNA isolated from whole blood (dFdC/dG ratio), as determined by liquid chromatography-mass spectrometry. Assessments were done by Advion BioSciences, Inc. Ithaca, NY, USA.
A validated Vendex Cell Search method was used to quantify the number of circulating endothelial cells (CEC) in blood samples (~10 mL).
Results
Patients
A total of 15 patients with various advanced solid tumors started combination treatment with LY2334737 (10–40 mg/day) and capecitabine (650 mg/m2 BID). The population included 13 Caucasian, 1 Hispanic, and 1 African patient (Table 1). All completed at least 1 treatment cycle; patients received a median of 2 and a maximum of 7 cycles. Fourteen patients discontinued treatment due to PD, and 1 patient due to toxicity (pyrexia).
Dose-limiting toxicities and general safety assessment
On the initial dose level of capecitabine (650 mg/m2 BID), LY2334737 dose levels of 10, 20, 25, 35, and 40 mg/day were explored. The MTD was not reached because the sponsor decided to stop enrollment after a cluster of unexpected, mainly hepatic toxicities had occurred in a study of Japanese patients on single-agent treatment with LY2334737 QD at doses ≥30 mg per day [10]. There were 2 patients with observed DLTs: 1 patient with G3 fatigue at 35 mg LY2334737 and 1 patient with G3 diarrhea and G3 hyponatremia at the 40 mg dose level.
No patient died while on study drug, but 2 patients died from disease within 30 days after the last dose. Serious AEs were reported by 5 patients, only 1 event (G3 diarrhea, patient on 40 mg dose) was considered related to study drug. The most frequent AEs (all grades) potentially related to study drug were fatigue (46.7 %), diarrhea (40.0 %), nausea (26.7 %), vomiting (26.7 %), and asthenia (26.7 %). No G3/4 hematologic toxicities (i.e., CTCAE related to study drug) were reported; thrombocytopenia was observed in a single patient only (G1). Four patients (26.7 %) experienced non-hematologic G3 CTCAE toxicities (palmar–plantar erythrodysesthesia syndrome, hyponatremia, fatigue, and diarrhea), there were no G4 toxicities. Regarding hepatic toxicities, 1 Caucasian patient on the 20 mg dose reported G2 transaminase elevations (aspartate transaminase [AST], alanine transaminase [ALT]) during Cycle 1. A second Caucasian patient on the 25 mg dose reported G2 elevation of alkaline phosphatase plus G1 elevations of AST and ALT during Cycle 1; this patient also had G1 thrombocytopenia during Cycle 1.
Tumor response
Seven of the 15 patients (46.7 %) achieved a best tumor response of stable disease (SD), lasting for ≥5 cycles in 3 patients. There were no partial or complete responses, 6 patients had PD, and 2 patients had no post-baseline radiological assessments.
Pharmacokinetics
Figure 1 shows plasma concentration profiles of LY2334737 and its active metabolite dFdC after a single dose as monotherapy (Day 2 of Cycle 1; data for different dose levels normalized to the 40 mg dose level), and the corresponding profiles of a single LY2334737 40 mg dose from the first-in-human Phase 1 Study, JLBA [4]. LY2334737 and dFdC plasma concentration profiles remained within the interval (10th to 90th percentile) predicted by Study JLBA. The parallel disposition patterns of LY2334737 and dFdC indicate that dFdC is a formation-rate-limited metabolite of LY2334737. Notable PK parameters for LY2334737 included moderate-to-high apparent clearance (CL/F; range 144–311 L/h) and a high apparent volume of distribution (V/F; range 359–854 L).

LY2334737 and dFdC concentration versus time profiles after a single dose of LY2334737 (Day 2, Cycle 1), data normalized to 40 mg dose. dFdC 2′,2′-difluorodeoxcytidine, hr hour, JLBA previous first-in-human Phase 1 study [4], SD standard deviation
Figure 2 shows the steady-state plasma concentration profiles of LY2334737 and dFdC after 2 weeks of combined treatment with LY2334737 QD and capecitabine 650 mg/m2 BID, in comparison to the steady-state profiles obtained in Study JLBA after 2 weeks of LY2334737 monotherapy at 40 mg per day. The PK profiles were similar and revealed no indication that combined administration with capecitabine might change the PK of LY2334737 or dFdC. The mean ratio (90 % CI) between the AUC in the presence and absence of concomitant capecitabine BID treatment was 1.38 (1.00, 1.77) for LY2334737 and 1.19 (0.28, 2.10) for dFdC. These values were within the observed variability range of LY2334737 observed in Study JLBA, where the coefficient of variation for the AUC of LY2334737 had been 36 % at the 40 mg dose [4].

LY2334737 and dFdC concentration versus time profiles after 2 weeks of treatment with LY2334737 QD and capecitabine BID (Day 15, Cycle 1), data normalized to 40 mg dose. dFdC 2′,2′-difluorodeoxcytidine, h hour, hr hour, JLBA previous first-in-human Phase 1 study [4], JLBC current study
Figure 3 shows the plasma concentration profiles of capecitabine and 5-FU after single dose administration of capecitabine as monotherapy (Cycle 1, Day 1), in combination with LY2334737 (Cycle 2, Day 1), and after 2 weeks of combined treatment (Cycle 1, Day 15; steady state). PK profiles were similar for both capecitabine and 5-FU at all 3 time points, indicating that co-administration of LY2334737 had no relevant impact on the PK of capecitabine and 5-FU. The mean ratio (90 % CI) between the AUCs measured in the presence and absence of LY2334737 was 1.00 (0.64, 1.36) for capecitabine and 1.29 (0.48, 2.10) for 5-FU, again revealing no indication drug interaction.

Capecitabine and 5-FU concentration versus time profiles after a single dose of capecitabine (Day 1, Cycle 1), in combination with LY2334737 (Day 1, Cycle 2), and after 2 weeks of combined treatment (Day 15, Cycle 1). 5-FU 5-fluorouracil, h hour. Values presented are means and standard deviations
Pharmacodynamic data
Figure 4 shows the incorporation of dFdC into the patient’s DNA isolated from whole blood, before and after 14 days of combined treatment with LY2334737 at doses of 10–40 mg/day and 650 mg/m2 capecitabine BID. Due to the small number of patients with data available, data for 10–25 mg and 35–40 mg LY2334737 dose groups were combined. A significant dose-dependent increase in the mean amount of dFdC incorporated into the DNA was observed (left panel). The majority of the dFdC/dG ratios observed in individual patients (right panel) fell within the spread of data observed in individual patients receiving the 40 mg dose in Study JLBA [4].

Amount of dFdC incorporated into DNA (extracted from whole blood), expressed as pg of gemcitabine per μg of deoxyguanosine (dFdC/dG). dFdC 2′,2′-difluorodeoxcytidine, dG deoxyguanosine, JLBA previous first-in-human Phase 1 study [4]; JLBC current study
Changes in the levels of CEC from baseline (Cycle 1, Day 1 pre-dose) after 2 weeks of combined treatment with LY2334737 and capecitabine were explored in 6 patients. CEC levels decreased in 1 patient with high baseline levels only (from 171 to 70 cells per 10 mL); 3 patients showed increases by >10 cells (+14, +30, +182 cells per mL).
Discussion
This Phase 1b study provides the first clinical and PK data for oral combination treatment with LY2334737 and capecitabine. Because the study was stopped early after 15 patients had been enrolled, the main objective, i.e., to determine the recommended Phase 2 dose, was not fully answered. No new or unexpected toxicities were observed, and the PK data revealed no indication for clinically relevant drug-drug interactions.
The current study was stopped early due to a cluster of unexpected toxicities that occurred in a separate Phase 1 study enrolling Japanese patients with single-agent LY2334737 [10]. At 40 mg/day, 3 patients experienced hepatic toxicities (G3/4 transaminase increase, G1-3 bilirubin increase, and several other hepatic events) and G4 thrombocytopenia during the first treatment cycle, and all 3 patients showed features of disseminated intravascular coagulation. A fourth patient experienced a G3 transaminase elevation at the 30 mg dose, but did not present with clinical features similar to the other 3 patients. All observed toxicities reverted to normal or near normal after drug discontinuation.
In contrast, doses up to 40 mg/day were tolerated without major hepatic toxicity in the first-in-human study of LY2334737 in 65 European patients [4]. Two cases of G3/4 thrombocytopenia were reported, both at a higher dose of 50 mg/day. In both cases liver involvement was minor, with G1 AST, ALT, and bilirubin elevations. These data are consistent with our study which included 13 Caucasian, 1 Hispanic, and 1 African patient. No G3/4 thrombocytopenia or hepatic toxicities were reported. One patient experienced G2 AST and ALT elevation but no thrombocytopenia, and a second patient experienced minor (G1) AST/ALT elevation, G2 alkaline phosphatase elevation and G1 thrombocytopenia. A comprehensive pharmacogenetic analysis of all trials with LY2334737 conducted to date is ongoing to identify potential reasons for the cluster of toxicities observed in Japanese, but not in Caucasian patients at the 40 mg dose level given QD.
In our study, no G3/4 hematologic toxicity and only 4 events of G3 non-hematologic toxicities (palmar-plantar erythrodysesthesia syndrome, hyponatremia, fatigue, diarrhea) were reported, indicating that the combination can be safely administered to patients with solid tumors using doses of 40 mg/day. The lack of G3/4 thrombocytopenia may be explained by the lower systemic exposure to gemcitabine over time that is provided by oral administration of LY2334737 in the dose range explored.
Nevertheless, the toxicity findings of the Japanese study indicate that QD administration of LY2334737 for 2 weeks, followed by 1 week of rest, may not offer the best risk/benefit ratio possible. The toxicity and efficacy of gemcitabine is known to highly depend on the schedule of administration [11–13]. Preliminary results from a Phase 1 study which explored alternative schedules in Caucasian patients indicate that low-dose, “metronomic” schedules, e.g., LY2334737 given every other day without drug-free interval, might be associated with less toxicity [14]. Larger studies with populations from different ethnicities are now in progress to identify the optimal dose and schedule for Phase 2 studies.
While efficacy was not a primary endpoint of this Phase 1b study, it was noted that 7 of the 15 patients treated (46.7 %) achieved SD. Similarly, “hints of anti-tumor activity or SD were observed in 22 of 51 evaluable patients” was seen in the JLBA study, with 2 patients achieving SD with progression–free survival [4].
The main limitation of the current study is that due to early termination, the sample size was small and no data could be obtained for higher capecitabine doses above 650 mg/m2 BID. But taken together, the PK information available indicates that there is no drug-drug interaction between LY2334737 and capecitabine, because (a) the PK profiles and AUCs after single doses of LY2334737 and capecitabine alone were similar to those after a single dose and after 14 days of combined treatment, (b) the PK profiles of LY2334737 when given alone or in combination with capecitabine were within the predicted range based on the previous data, (c) incorporation of the active metabolite dFdC into whole blood DNA throughout 14 days of combination treatment also was in the same range as observed in Study JLBA.
In conclusion, this study indicates that no PK interaction exists between LY2334737 and capecitabine when administered to patients with advanced/metastatic solid tumors. The recommended dose for Phase 2 studies could not be determined, but no unexpected toxicities were observed with LY2334737 doses up to 40 mg/day.
References
Pratt SE, Durland-Busbice S, Shepard RL, Heinz-Taheny K, Iversen PW, Dantzig AH (2013) Human carboxylesterase-2 hydrolyzes the prodrug of gemcitabine (LY2334737) and confers prodrug sensitivity to cancer cells. Clin Cancer Res 19:1159–1168
Gemzar Summary of Product Characteristics (2011) Available at: http://www.medicines.ie/medicine/8346/SPC/Gemzar/. Accessed on 11 February 2013
Koolen SL, Witteveen PO, Jansen RS, Langenberg MH, Kronemeijer RH, Nol A, Garcia-Ribas I, Callies S, Benhadji KA, Slapak CA, Beijnen JH, Voest EE, Schellens JH (2011) Phase I study of oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin Cancer Res 17:6071–6082
Pratt SE, Durland-Busbice S, Shepard RL, Donoho GP, Starling JJ, Wickremsinhe ER, Perkins EJ, Dantzig AH (2013) Efficacy of low-dose oral metronomic dosing of the prodrug of gemcitabine, LY2334737, in human tumor xenografts. Mol Cancer Ther 12:481–490
Le Tourneau C, Lee JJ, Siu LL (2009) Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101:708–720
Xeloda Summary of Product Characteristics (2014) Available at: https://www.medicines.org.uk/emc/medicine/4619. Accessed 21 October 2014
Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, Harper PG, Dunn J, Tudur-Smith C, West J, Falk S, Crellin A, Adab F, Thompson J, Leonard P, Ostrowski J, Eatock M, Scheithauer W, Herrmann R, Neoptolemos JP (2009) Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 27:5513–5518
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92:205–216
Veltkamp SA, Hillebrand MJ, Rosing H, Jansen RS, Wickremsinhe ER, Perkins EJ, Schellens JH, Beijnen JH (2006) Quantitative analysis of gemcitabine triphosphate in human peripheral blood mononuclear cells using weak anion-exchange liquid chromatography coupled with tandem mass spectrometry. J Mass Spectrom 41:1633–1642
Yamamoto N, Nokihara H, Yamada Y, Uenaka K, Sekiguchi R, Makiuchi T, Slapak CA, Benhadji KA, Tamura T (2013) Phase I study of oral gemcitabine prodrug (LY2334737) in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 71:1645–1655
Grunewald R, Kantarjian H, Keating MJ, Abbruzzese J, Tarassoff P, Plunkett W (1990) Pharmacologically directed design of the dose rate and schedule of 2′,2′-difluorodeoxycytidine (Gemcitabine) administration in leukemia. Cancer Res 50:6823–6826
Patel SR, Gandhi V, Jenkins J, Papadopolous N, Burgess MA, Plager C, Plunkett W, Benjamin RS (2001) Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol 19:3483–3489
Tempero M, Plunkett W, Ruiz Van Haperen V, Hainsworth J, Hochster H, Lenzi R, Abbruzzese J (2003) Randomized phase II comparison of dose-intense gemcitabine: 30-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 21:3402–3408
Faivre SJ, Olszanski AJ, Weigang-Koehler K, Riess H, Peng G, Callies S, Benhadji KA, Raymond E (2012) Phase I and pharmacokinetic (PK)/pharmacodynamic (PD) study of LY2334737, an oral gemcitabine prodrug, in patients (pts) with advanced solid tumors. Abstract. J Clin Oncol 30(suppl): abstr 2554
Acknowledgments
The study has been funded by Eli Lilly and Company. We would like to thank all patients for participating in the study. Karin Helsberg and Michael Riley, Trilogy Writing & Consulting, Frankfurt, Germany, provided medical writing support on behalf of Eli Lilly and Company.
Disclosure of potential conflicts of interest
The study was funded by Eli Lilly and Company. KAB and SC are employees of Eli Lilly and Company. JRI, GKD, GF, WWM, JB, and AAA have no no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Infante, J.R., Benhadji, K.A., Dy, G.K. et al. Phase 1b study of the oral gemcitabine ‘Pro-drug’ LY2334737 in combination with capecitabine in patients with advanced solid tumors. Invest New Drugs 33, 432–439 (2015). https://doi.org/10.1007/s10637-015-0207-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0207-9