
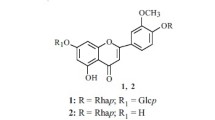
A new flavonol glycoside, 8-methoxykaempferol-3-O-α-L-rhamnosyl-4′-O-β-D-glucoside (1), was isolated from the fruits of Cercidiphyllum japonicum (Cercidiphyllaceae) in addition to two known anomeric galloyltannins, [2,2′,5-tri-O-galloyl-α/β-D-hamamelose (2) and 3,4,6-tri-O-galloyl-α/β-D-glucose (3)], and one known flavonol [quercetin (4)]. The structures of the isolated compounds were elucidated using chemical and spectral methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cercidiphyllum japonicum Siebold & Zucc., also known as katsura, is a flowering tree species of Cercidiphyllum, the sole genus in the family Cercidiphyllaceae [1, 2]. C. japonicum, the deciduous tree, was well represented in the fossil record, occurring in the late Cretaceous and Tertiary of Europe and North America [2]. But the tree is now listed as an endangered species in China and only confined to eastern Asia regions, such as China, Japan, as well as the Korean peninsula [2,3,4]. C. japonicum can grow up to 10–45 m tall, with a trunk diameter of approximately 2 m. Its flowers are inconspicuous, blooming in early spring among the opening leaves, with female and male flowers on separate plants (dioecious) [5]. The fruit of C. japonicum is a cluster of two to four follicles 1–1.8 cm long and 2–3 mm wide, each follicle containing several winged seeds [6, 7].
C. japonicum has been widely used in traditional medicine for its antifungal, antioxidative, anti-inflammatory, antimicrobial, and hair growth promoting effects, as well as the bioactivities of arresting convulsion and subduing liver wind [8,9,10]. Previous phytochemical investigations revealed that chemical constituents such as kaempferol-7-O-β-glucoside, quercetin-3-O-β-glucoside, kaempferol-3-O-β-glucoside, and 8-methoxykaempferol-3-O-β-glucoside in wood extracts of C. japonicum bear anti-ice nucleation activity that promotes supercooling of water [11]. Our earlier phytochemical investigations on leaves and barks of C. japonicum resulted in the isolation and purification of several phenolics, such as tannins, phenolic acids, and flavonoids [1, 4, 12,13,14]. However, up to now, no investigation has been carried out to study the secondary metabolites of C. japonicum fruits. In the current work, we report the extraction, separation, and structural elucidation of a new flavonol glycoside, 8-methoxykaempferol-3-O-α-L-rhamnosyl-4′-O-β-D-glucoside (1), along with two known anomeric galloyltannins, [2,2′,5-tri-O-galloyl-α/β-D-hamamelose (2) and 3,4,6-tri-O-galloyl-α/β-D-glucose (3)], and one known flavonol [quercetin (4)] [15,16,17,18], from the fruits of C. japonicum for the first time.
Compound 1 was isolated as a yellowish amorphous powder with optical rotation \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –21.2° (c 0.5, MeOH) and mp 239–241°C (uncorrected). The molecular formula of compound 1 was deduced to be C28H32O16 by its FAB-MS spectra (positive mode) for [M + K]+, [M + Na]+, and [M + H]+ ion peaks found at m/z 663, 647, and 625, respectively.
The presence of one or more phenolic hydroxyl groups in compound 1 was confirmed by the dark green color in a TLC experiment when spraying with 1% FeCl3 (in EtOH) solution followed by heating [19]. In 2D TLC, compound 1 gave Rf values of 0.90 and 0.47 when solvents A and B were used as the developing solvents, respectively. In the IR spectrum, compound 1 displayed characteristic absorption bands at 3400 cm–1 (OH), 1632 (α,β-unstaturated C=O), 1590, 1510, 1490 cm–1 (aromatic C=C), and 1160, 1060, 1025 cm–1 (C–O) [20]. The UV spectrum of 1 showed absorption bands at λmax 225, 275, 325 and 355 nm (in MeOH), coinciding well with a 3-O-glycoside of 8-substitution flavonol structure [21].
In the 1H NMR spectrum of compound 1 (Table 1), a singlet resonating at δ 6.29 was attributable to H-6 of the flavonoid A-ring. Another singlet found at δ 12.25 was assigned to the free hydroxyl group at C-5. A pair of AA′BB′ system proton signals consisting of two doublets (J = 8.0 Hz) at δ 7.10 and 8.12 was ascribed to H-3′, 5′ and H-2′, 6′ due to the para-substituted B-ring [22]. A methoxy group was recognized by its 13C and 1H NMR peaks [δC 60.6, δH 3.83 (3H, s)], and its connection to C-8 was confirmed by the HMBC spectrum for the 1H→13C coupling correlation was observed between δH 3.83 (3H, s) of the methoxy group and C-8 (δC 127.7), as shown in Fig. 1. In the 13C NMR spectrum of 1, characteristic flavonol signals were observed at δ 156.1 (C-2), 133.7 (C-3), and 177.6 (C-4). Therefore, the aglycone of compound 1 was elucidated as 8-methoxykaempferol [21].

Key HMBC correlations observed in compound 1.
In the 1H NMR spectrum, an anomeric proton typically rotated at δ 4.65 (1H, d, J = 1.7, H-1′′), together with a secondary methyl group characteristically centered at δ 1.03 (3H, d, J = 6.5 Hz, H-6′′), indicating the presence of an α-configuration of L-rhamnose in compound 1. In the 13C NMR spectrum, the down-field (Δδ ~10 ppm) shift of C-2 (δC 156.1) in compound 1 compared with that in kaempferol confirmed that the glycosylation appeared at 3-hydroxy of the aglycone [23]. The linkage of the α-L-rhamnosyl moiety at 3-OH of 8-methoxykaempferol was additionally verified by the HMBC data of compound 1, where the long-range coupling correlation was observed from H-1′′ (δ 4.65, d, J = 1.7) of the rhamnosyl residue to C-3 (δ 133.7) [23].
The presence of a β-D-glucosyl unit in compound 1 was confirmed by its NMR data: an anomeric proton signal as a doublet with coupling constant J = 7.3 Hz rotated at δ 5.11 (1H, d, H-1′′′) in the 1H NMR spectrum, and six carbon signals were observed at δ 101.1, 74.3, 77.5, 70.1, 77.2, and 61.3 in the 13C NMR spectrum [24]. The HMBC spectroscopic correlations of 1, as displayed in Fig. 1, showed interlinks between the anomeric proton H-1′′′ (δ 5.11, d, J = 7.3 Hz) and C-4′ (δ 160.9) of the aglycone B-ring, which indicated that the glucosyl moiety was connected to 4′-OH of 8-methoxykaempferol.
Based on the data and analysis above, compound 1 was structurally elucidated as 8-methoxykaempferol-3-O-α-Lrhamnosyl-4′-O-β-D-glucoside, which is a new compound.
The structures of the three known compounds, including two anomeric galloyltannins (2 and 3) and one flavonol (4), were determined by comparing their NMR, MS, UV, and IR values with those in the literature [15,16,17,18]. To the best of our knowledge, 3,4,6-tri-O-galloyl-α/β-D-glucose (3) has never previously been extracted from any plant in Cercidiphyllaceae family.
Experimental
General. IR spectra were obtained on a PerkinElmer BX FT infrared spectrometer by the KBr disk method. UV absorbancies were determined with a Jenway 6405 ultraviolet spectrophotometer. Optical rotation (in MeOH) was measured on a JASCO DIP-1000 digital polarimeter. Melting point (uncorrected) was determined using an Electro Thermal 9100 apparatus. Positive FAB-MS spectra were obtained on a Micromass Autospec M363 spectrometer. 1H and 13C NMR spectra at a frequency of 600 and 150 MHz, respectively, were recorded in DMSO-d6 using tetramethylsilane as internal standard on a Bruker Avance III 600 spectrometer. Column chromatography was carried out using packing materials of Sephadex LH-20 and silica gel. TLC experiments were performed on DC-Plastikfolien Cellulose F plates with t-BuOH–AcOH–H2O (3:1:1, solvent A) and AcOH–H2O (3:47, solvent B) used as developing solvents. TLC visualization was conducted by UV light (254 & 365 nm) and by spraying with 1% ethanolic FeCl3 followed by heating for 15–60 seconds.
Plant Material. Fruits of C. japonicum were collected in October of 2016 from Xuchang region of Henan Province, China, and the plant materials were identified by Prof. Y. S Bae at the Department of Forest Biomaterials Engineering, College of Forest & Environment Sciences, Kangwon National University, Korea. A voucher specimen (No. 20161001) has been deposited in the Herbarium of Tianjin Key Laboratory of Pulp and Paper, College of Papermaking Science and Technology, Tianjin University of Science and Technology, China.
Extraction and Isolation. Fruits of C. japonicum (3.97 kg) were air-dried, finely ground, then extracted four times (more than 72 h each time) with acetone–H2O (7:3) mixture at room temperature. After combination and evaporation under reduced pressure, the crude residues were diluted with H2O and sequentially subjected to liquid–liquid fractionation using n-hexane, CHCl3, EtOAc, and n-BuOH in separation funnels, then freeze dried to give n-BuOH fraction powders (labeled as FCJB). A portion of FCJB (22.07 g) was subjected to silica gel column chromatography using gradient mixtures of CHCl3–acetone (4:1→1:19) to give seven fractions FCJB1–FCJB7. Fraction FCJB2 (9.06 g) was successively separated by Sephadex LH-20 column chromatography with MeOH–H2O (4:1, 2:1, 1:3) and n-hexane–EtOH (1:1, 1:3) as eluents to yield compounds 2 (37.1 mg), 3 (52.6 mg), and 4 (71.4 mg). Fraction FCJB5 (7.15 g) was further purified by silica gel column chromatography using CHCl3–acetone (99:1→1:1) as mobile solvents to get three subfractions FCJB51–FCJB53. Subfraction FCJB52 was then subjected to Sephadex LH-20 column chromatography with MeOH–H2O (2:1, 1:3, 1:5) as eluting mixture to give four fractions FCJB521–FCJB524. Fraction FCJB523 was repeatedly subjected to column chromatography packed with Sephadex LH-20 and washed with n-hexane–EtOH (1:1, 1:4) to obtain 40.5 mg of compound 1.
8-Methoxykaempferol-3-O-α-L-rhamnosyl-4′-O-β-D-glucoside (1). Yellowish amorphous powder, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –21.2° (c 0.5, MeOH); mp 239–241°C. IR (KBr, νmax, cm–1): 3400, 1632, 1590, 1510, 1490, 1160, 1060, 1025. UV (MeOH, λmax, nm): 225, 275, 325, 355; Rf 0.90 (solvent A) and 0.47 (solvent B). FAB-MS m/z: 663 [M + K]+, 647 [M + Na]+, and 625 [M + H]+, corresponding to molecular weight 624 and calculated for C28H32O16. For 1H and 13C NMR data, see Table 1. For key HMBC correlations, see Fig. 1.
References
M. S. Lee, H. J. Min, C. L. Si, and Y. S. Bae, J. Korean Wood Sci. Technol., 44 (4), 559 (2016).
S. D. Sarker, P. Whiting, R. Lafent, J. P. Girault, and L. Dinan, Biochem. Syst. Ecol., 25, 79 (1997).
Y. Isagi, M. Kudo, K. Osumi, T. Sato, and H. Sakio, Mol. Ecol. Notes, 5, 596 (2005).
Y. S. Bae, H. J. Min, M. S. Lee, and Y. K. Kim, J. Korean Wood Sci. Technol., 45 (3), 250 (2017).
P. Teifel and R. G. Berger, J. Sci. Food Agric., 63, 59 (1993).
M. Tada and K. Sakurai, Phytochemistry, 30 (4), 1119 (1991).
D. H. Wang, J. Kasuga, C. Kuwabara, K. Endoh, Y. Fukushi, S. Fujikawa, and K. Arakawa, Planta, 235, 747 (2012).
Zijiang Li, Jiaxin Yang, Hui Wang, Rui Xu, Zetong Rao, Chuan-Ling Si, Jie Zhang, Lin Sun, Xiaoyi Zhang, Shujun Han, Zhe Sun, Lei Wu, Dan Liu, Ying Liu, and Jun-Hui Wang, Chem. Nat. Compd., 55, 121 (2019).
M. Takasugi and N. Katui, Phytochemistry, 25 (12), 2751 (1986).
K. Towatari, K. Yoshida, N. Mori, K. Shimizu, R. Kondo, and K. Sakai, Planta Med., 68, 995 (2002).
J. Kasuga, Y. Hashidoko, A. Nishioka, M. Yoshiba, K. Arakawa, and S. Fujikawa, Plant Cell Environ., 31 (9), 1335 (2008).
T. S. Lee and Y. S. Bae, J. Korean Wood Sci. Technol., 43 (5), 558 (2015).
M. S. Lee, H. J. Min, J. K. Kim, and Y. S. Bae, J. Korean Wood Sci. Technol., 44 (4), 551 (2016).
T. S. Lee, H. J. Lee, and Y. S. Bae, J. Korean Wood Sci. Technol., 43 (5), 591 (2015).
M. S. Lee, H. J. Min, C. L. Si, and Y. S. Bae, J. Korean Wood Sci. Technol., 44 (4), 559 (2016).
E. A. Haddock, R. K. Gupta, S. M. K. Al-Shafi, E. Haslam, and D. Magnolato, J. Chem. Soc. Perkin Trans. 1, 2515 (1982).
L. Wu, G. C. Wang, T. Shen, Q. Qiang, Q. Xue, M. Chen, J. M. Zhang, Y. Y. Luo, Y. M. Hong, C. L. Si, and W. C. Hu, Chem. Nat. Compd., 54, 210 (2018).
B. Ternai and K. R. Markham, Tetrahedron, 32, 565 (1976).
C. L. Si, D. N. Xie, B. Sun, J. H. Wang, C. Y. Liu, L. L. An, W. Q. Zhou, Z. Y. Tao, and Y. M. Hong, Chem. Nat. Compd., 53, 866 (2017).
L. Q. Hu, K. Wang, G. B. Li, R. Y. Zhang, Y. Y. Luo, C. L. Si, and J. H. Wang, Holzforschung, 71, 785 (2017).
J. C. Dauguet, M. Bert, J. Dolley, A. Bekaert, and G. Lewin, Phytochemistry, 33, 503 (1993).
F. B. C. Okoye, W. R. Sawadogo, J. Sendker, A. H. Aly, B. Quandt, V. Wray, A. Hensel, C. O. Esimone, A. Debbab, and M. Diederich, J. Ethnopharmacol., 176, 27 (2015).
C. Y. Liu, D. N. Xie, L. L. An, Z. J. Li, C. L. Si, Y. S. Bae, G. Xu, and L. Wu, Chem. Nat. Compd., 53, 1020 (2017).
C. L. Si, S. Fan, and L. L. An, Chem. Nat. Compd., 52, 1008 (2016).
Acknowledgment
The authors thank the Foundations of State Key Laboratory of Tree Genetics and Breeding (No. K2017101) and Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, Guangxi University (No. KF201801-5).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2019, pp. 217–219.
Rights and permissions
About this article

Cite this article
Si, CL., Chen, S., Li, Z. et al. A New Flavonol Glycoside from the Fruits of Cercidiphyllum japonicum. Chem Nat Compd 55, 252–255 (2019). https://doi.org/10.1007/s10600-019-02661-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-019-02661-3