
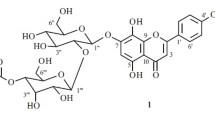
Phytochemical investigation of the n-BuOH fraction of the 70% EtOH extract of Juglans sigillata green husks afforded a new isoflavone triglycoside, named 3′-methoxy-5′-hydroxy-isoflavone-7-O-(2″,6″-di-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside (1), along with two known epimeric ellagitannins, pedunculagin (2) and 2,3-O-4,4′,5,5′,6,6′-hexahydroxydiphenoyl-(α/β)-glucose (3). Their structures were elucidated on the basis of spectroscopic analysis and chemical evidence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The genus Juglans, belonging to the plant family Juglandaceae, is distributed widely in the temperate and subtropical regions of the world [1]. J. sigillata Dode is indigenous to China and has been extensively cultivated in mountain slopes and valleys of Tibet, Yunnan, Guizhou, and Sichuan Provinces of southwest China for its edible and highly nutritional nuts [2, 3]. Green husks of J. sigillata, commonly known as Qing-Long-Yi, have long been used in Chinese traditional medicine owing to its anti-inflammatory, antitumor, anti-oxidative, and antinociceptive properties [1, 3]. Previous phytochemical study of J. sigillata green husks led to the isolation of flavan-3-ols, galloylglucosides, sinapaldehyde, (Z)-10-eicosenoic acid, naphthoquinones, and α-tetralone derivatives [1, 3,4,5]. In the current work, repeated column chromatography of the n-BuOH soluble fraction of the 70% EtOH extract resulted in the isolation of a new isoflavone triglycoside, 3′-methoxy-5′-hydroxyisoflavone-7-O-(2″,6″-di-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside (1) (Fig. 1) with molecular formula C34H42O18, as well as two known epimeric ellagitannins, pedunculagin (2) and 2,3-O-4,4′,5,5′,6,6′-hexahydroxydiphenoyl-(α/β)-glucose (3).


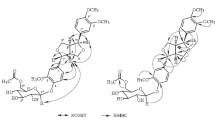
Key HMBC correlations observed in compound 1.
Compound 1 was isolated as a pale amorphous powder with melting point 174–176°C and optical rotation \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –23.2° (c 0.5; MeOH). Its molecular formula was determined as C34H42O18 by positive FAB-MS spectrometric analysis, for its [M + H]+ ion peak appeared at m/z 739. The phenolic hydroxyl group in compound 1 was confirmed from its TLC chromogenic reaction by the gray-green color when spraying with ferric chloride reagent (R f values 0.22 and 0.80 in solvents A and B, respectively) [6, 7]. IR absorption bands of compound 1 were found at 1645 cm–1 (isoflavone carbonyl group) and 3410 cm–1 (hydroxyl group) [8].
Compound 1 was recognized as an isoflavone derivative by its UV absorption maxima at 211, 255, and 295 nm [9], and by the characteristic proton singlet at δ 8.35 (H-2) in the 1H NMR spectrum [8, 10]. For compound 1, its 1H NMR spectrum (Table 1) displayed the presence of a pair of classical ABX type proton signals [δ 7.99 (1H, d, J = 8.6 Hz), δ 7.10 (1H, dd, J = 1.9, 8.6 Hz), and δ 7.18 (1H, d, J = 1.9 Hz)], which were assignable to H-5, H-6, and H-8, respectively. Three proton signals observed at δ 7.02 (1H, d, J = 1.7 Hz), 7.07 (1H, t, J = 1.7 Hz), and 7.01 (1H, t, J = 1.7 Hz) were ascribed to aromatic H-2′, H-4′, and H-6′, respectively [11]. The methoxyl protons of H-7′ resonating as a singlet were characterized at δ 3.79. The 1H NMR spectrum also revealed the existence of three sugar moieties in compound 1: an anomeric proton of β-D-glucose at δ 5.16 (1H, d, J = 7.5 Hz), two secondary methyls of α-L-rhamnoses at δ 1.35 (3H, d, J = 6.1 Hz) and 1.18 (3H, d, J = 6.3 Hz), and two anomeric protons of α-L-rhamnoses at δ 4.68 (1H, d, J = 1.5 Hz) and 5.26 (1H, d, J = 1.6 Hz), respectively [12, 13].
In the 13C NMR spectrum, carbon signals at δ 153.58 (C-2) and δ 174.90 (C-4) also confirmed that compound 1 was an isoflavone derivative and no hydroxyl was attached to its C-5 [11, 14].
As summarized in Table 1, the heteronuclear multiple bond correlation (HMBC, Fig. 1) experiment results of compound 1 showed interlinks between the glucose anomeric H-1″ (δ 5.16, 1H, d, J = 7.5 Hz) and C-7 (δ 161.71), which indicated that the glucose is attached to C-7 of the isoflavone aglycone. Additionally, the cross correlations observed in HMBC between one rhamnose anomeric H-1‴ (δ 4.68, 1H, d, J = 1.5 Hz) and C-6″ (δ 67.88), between another rhamnose anomeric H-1′′′′ (δ 5.26, 1H, d, J = 1.6 Hz) and C-2″ (δ 78.90), and between H-7′ (δ 3.79, 3H, s) and C-3′ (δ 145.78) confirmed that the methoxyl group is linked to the C-3′ site of the aglycone, and the two α-L-rhamnosyl residues are connected to C-6″ and 2″ of the β-D-glucosyl moiety, respectively. Thus, compound 1 was determined as an isoflavone triglycoside, and its structure was elucidated as 3′-methoxy-5′-hydroxyisoflavone-7-O-(2″,6″-di-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside.
Experimental data of distortionless enhancement by polarization transfer (DEPT) spectrum, as demonstrated in Table 1, classified 34 carbon peaks of compound 1 into eight quaternary (C), 22 methine (CH), one methylene (CH2), and three methyl (CH3) carbon signals, with each signal in good agreement with the corresponding sites of 3′-methoxy-5′-hydroxy-isoflavone-7-O-(2″,6″-di-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside.
The chemical structures of the two known epimeric ellagitannins, pedunculagin (2) and 2,3-O-4,4′,5,5′,6,6′-hexahydroxydiphenoyl-(α/β)-glucose (3), were identified and established through spectroscopic evidence and by comparison with previously published data [15, 16]. To the best of our knowledge, this is the first report of compounds 2 and 3 from green husks of J. sigillata.
Experimental
General Methods and Equipment. NMR experiments were carried out in DMSO-d6 with SiMe4 as an internal standard on a Bruker Avance DPX 400 spectrometer at frequencies of 400 and 100 MHz for 1H and 13C NMR, respectively. IR spectra were obtained in KBr disks on a PerkinElmer BX FT-IR spectrometer, and UV spectra were recorded on a Jenway 6405 spectrophotometer. FAB-MS (positive) spectra were obtained on a Micromass Autospec M363 spectrometer. A Jasco DIP-1000 digital polarimeter was used to record the optical rotations. Melting points were determined with an Electro Thermal 9100 apparatus and were uncorrected. Silica gel (Shanhai Anyan Biological Company, China) and Sephadex LH-20 (Sigma, USA) were used in open column chromatography. Fraction eluents were collected with BS-100A fraction collectors. In thinlayer chromatography (TLC) experiments, DC-Plastikfolien Cellulose F (Merck Co.) plates were used with t-BuOH–AcOH–H2O (3:1:1, solvent A) and AcOH–H2O (3:47, solvent B) as developing solvents. TLC spot detection was performed by UV light (365 and 254 nm) and by spraying with 1% ethanolic FeCl3 solution followed by heating.
Plant Material. Green husks of J. sigillata were obtained in Santai County, Sichuan Province of China in August 2009 and identified by Prof. Dan Wang at the Institute of the Chemical Industry of Forest Products, China. A voucher specimen (No. 200908001) has been deposited in the Herbarium of Tianjin Key Laboratory of Pulp and Paper, College of Papermaking Science and Technology, Tianjin University of Science and Technology, China.
Extraction and Purification. Air-dried and finely ground green husks of J. sigillata were soaked in 70% EtOH for more than 72 h. After filtration, concentration, and removal of EtOH under reduced pressure, the extract was suspended in water and sequentially fractionated with n-hexane, CH2Cl2, EtOAc, and n-BuOH, then freeze dried. A portion of the resulting n-BuOH fraction (29.58 g) was subjected to open column chromatography packed with silica gel and a gradient of EtOAc–H2O–MeOH (28:1:1→18:1:1→8:1:1→3:1:1) used as the washing solvents system to give five fractions, named as JSGHB-1 (0.89 g), JSGHB-2 (5.16 g), JSGHB-3 (2.28 g), JSGHB-4 (16.85 g), and JSGHB-5 (1.54 g), which were grouped and their compositions monitored by TLC performance. Fraction JSGHB-4 was further loaded onto a Sephadex LH-20 column using MeOH–H2O (3:1) as an eluting solvent to obtain four fractions (JSGHB -41 to JSGHB -44). Fraction JSGHB-42 (8.96 g) was also subjected to open column chromatography with Sephadex LH-20 and n-hexane–EtOH (1:2) used as stationary phase and mobile solvents, respectively, to give five fractions (JSGHB-421 to JSGHB-425). Fraction JSGHB-423 (2.92 g) was successively chromatographed with Sephadex LH-20 open columns using MeOH–H2O (1:2 and 1:4) and n-hexane–EtOH (2:1 and 5:1) to give two amorphous powders: 40.6 mg of compound 1 and 106.0 mg of compound 2. Fraction JSGHB-424 (2.15 g) was also loaded onto a Sephadex LH-20 column with MeOH–H2O (1:1 and 1:3) and n-hexane–EtOH (3:1) as washing eluents to get 175.6 mg of compound 3.
3′-Methoxy-5′-hydroxyisoflavone-7- O -(2″,6″-di- O - α -L-rhamnopyranosyl)- β -D-glucopyranoside (1). Pale amorphous powders; mp 174–176°C; \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –23.2° (c 0.5; MeOH). IR (KBr, νmax, cm–1): 1645 (isoflavone carbonyl group) and 3410 (hydroxyl group). UV (MeOH, λmax, nm): 211, 255, and 295. R f 0.22 (solvent A) and 0.80 (solvent B). Positive FAB-MS m/z [M + H]+ at 739, suggested molecular weight 738 and calculated for C34H42O18; 1H and 13C NMR spectroscopic data in DMSO-d6 are shown in Table 1.
References
Q. Liu, P. Zhao, X. C. Li, M. R. Jacob, C. R. Yang, and Y. J. Zhang, Helv. Chim. Acta, 93, 265 (2010).
Z. Y. Wu and P. H. Raven, Flora of China, Vol. 4, Science Press, Beijing, 1999, p. 283.
C. L. Si, Y. Zhang, Z. Y. Zhu, and S. C. Liu, Chem. Nat. Compd., 47, 442 (2011).
D. M. Li, Y. L. Peng, and G. M. Li, Chin. Trad. Herb. Drug., 46, 962 (2015).
C. L. Si, P. P. Qin, H. Y. Hu, J. Z. Jiang, and Y. H. Ni, J. Biobased Mater. Biol., 5, 288 (2011).
C. L. Si, G. H. Xu, X. F. Huang, Z. G. Du, L. Wu, and W. C. Hu, Chem. Nat. Compd., 52, 132 (2016).
C. L. Si, G. J. Yu, Z. G. Du, X. F. Huang, S. Fan, H. S. Du, and W. C. Hu, Holzforschung, 70, 39 (2016).
D. K. Semwal, U. Rawat, R. Semwal, R. S. Rhhiya, P. Krishaw, and M. Singh, J. Asian Nat. Prod., 11, 1045 (2009).
V. Rukachaisirikul, Y. Sukpondma, C. Jansakul, and W. C. Taylor, Phytochemistry, 60, 827 (2002).
Y. M. Kang and N. H. Lee, Bull. Korean Chem. Soc., 32, 1048 (2011).
Z. Z. Cao, Y. Cao, Y. J. Yi, Y. P. Wu, Z. K. Leng, L. Du, and N. L. Owen, Acta Pharm. Sin., 34, 392 (1999).
C. L. Si, L. L. An, D. N. Xie, C. Y. Liu, X. Q. Chen, G. H. Wang, D. Huo, Q. L. Yang, and Y. M. Hong, Wood Sci. Technol., 50, 645 (2016).
C. L. Si, J. Z. Jiang, S. C. Liu, H. Y. Hu, X. D. Ren, G. J. Yu, and G. H. Xu, Holzforschung, 67, 357 (2013).
N. I. Kulesh, O. B. Maksimov, V. A. Denisenko, and V. P. Glazunov, Chem. Nat. Compd., 37, 29 (2001).
T. Tanaka, H. Tachibana, G. Nonaka, I. Nishioka, F. L. Hsu, H. Kohda, and O. Tanaka, Chem. Pharm. Bull., 41, 1214 (1993).
M. K. Seikel and W. E. Hillis, Phytochemistry, 9, 1115 (1970).
Acknowledgment
This research was funded by the Open Fund of State Key Laboratory of Tree Genetics and Breeding (Chinese Academy of Forestry) (Grant No. TGB2016002), Foundation of Jiangsu Provincial Key Laboratory of Pulp and Paper Science and Technology (201531), Nanjing Forestry University, China, State Key Laboratory of Pulp and Paper Engineering (South China University of Technology) (201611, 201211, 201451), National Natural Science Foundation of China (31501440).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2017, pp. 737–739.
Rights and permissions
About this article

Cite this article
Si, CL., Xie, DN., Sun, B. et al. A New Isoflavone Triglycoside from Green Husks of Juglans sigillata . Chem Nat Compd 53, 866–869 (2017). https://doi.org/10.1007/s10600-017-2142-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-2142-9