
A new diterpenoid, 19-O-β-D-glucopyranosyl-labda-8(17),14-dien-13-ol (1), and nine known triterpenes (2–10) were isolated from the aerial parts of Tripterygium hypoglaucum. All structures were elucidated by NMR spectroscopic methods. Moreover, the inhibition of superoxide anion generation and elastase release were also examined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The genus Tripterygium (Celastraceae) includes three species: Tripterygium wilfordii Hook. f., Tripterygium hypoglaucum (Levl.) Hutch, and Tripterygium regelii Sprague et Takeda [1]. The principal chemical constituents of Tripterygium are diterpene, triterpene, and sesquiterpene alkaloids, and there are also flavonoids, steroids, tannins, and glucides in it [2]. A number of terpenoids isolated from Tripterygium plants, which modulate the production of interferon gamma (IFN-γ), IL-1β, 2, 4, 8, and TNF-α[3,4,5,6,7], exhibit anti-inflammatory and immunosuppressive activities. T. hypoglaucum and T. wilfordii, two morphologically similar species from this genus, have been used for many years in traditional Chinese medicine for the treatment of swelling, inflammation, cancer, rheumatoid arthritis, and as insecticides [8]. In order to discover more structurally interesting and anti-inflammatory secondary metabolites from the genus Tripterygium, a phytochemical investigation on T. hypoglaucum was carried out, which led to the isolation of a new diterpenoid and nine known triterpenes. In this paper, we described the isolation and structural determination of the isolates, as well as their anti-inflammatory activity.
Compound 1 was obtained as a colorless oil, and its molecular formula C26H44O7 was established on the basis of HR-EI-MS (m/z 468.3108 [M]+; calcd 468.3087), indicating five degrees of unsaturation. The 1H and 13C NMR spectra of 1 (Table 1) showed characteristic resonances of a sugar and a diterpene moiety. The acid hydrolysis of 1 with 1 M HCl indicated the D-glucose as the sugar residue, which was determined by GC analysis of its corresponding trimethylsilylated L-cysteine derivative. The resonance due to the anomeric proton of the glycoside at δ 4.87 (d, J = 6.5 Hz) suggested the presence of a β-glucose moiety [9]. Except for the signals of the β-D-glucose, three tertiary methyl groups [δH 1.49, 1.16, 0.66 (each 3H, s); δC 28.4, 28.5, 15.6 (each, q)], ten methylenes including an oxygenated [δH 4.00 (1H, d, J = 8.0 Hz, Ha-19), 3.89 (1H, d, J = 8.0 Hz, Hb-19); δC 72.8 (t, C-19)], and two unsaturated ones [δH 5.58 (1H, dd, J = 14.4, 1.6 Hz, Ha-15), 5.16 (1H, dd, J = 8.9, 1.6 Hz, Hb-15), 4.90 (1H, br.s, Ha-17), 4.81 (1H, br.s, Hb-17); δC 111.1 (t, C-15), 107.2 (t, C-17)], two saturated and one olefinic [δH 6.20 (1H, dd, J = 14.4, 8.9 Hz, H-14); δC 147.5 (d, C-14)] methines, as well as four quaternary carbons including an oxygenated one [δC 72.7 (s, C-13)] and an unsaturated one [δC 148.9 (s, C-8)], were observed in the 1H and 13C NMR spectra, which were assigned to the labdane diterpene skeleton [10].

Careful comparison of the NMR data of 1 with those of (4R,13S)-18-O-β-D-glucopyranosyl-labda-8(17),14-dien-13-ol [11] showed that they were extremely similar, except that the chemical shifts at δ 49.3 (C-5), 79.3 (C-18), and 18.0 (C-19) in the reference compound were changed to δC 56.6, 28.5, and 72.8, respectively, in 1. These differences suggested that the β-D-glucose was attached to C-19 in 1 rather than C-18. The HMBC correlations (Fig. 1) of 1 observed between the anomeric proton (H-1′) and the oxygenated methylene carbon (C-19), and the ROESY correlations (Fig. 1) of H3-20/H2-19, H-5/H3-18, and H-5/H-9 confirmed the location of the oxygenated methylene at C-19. Based on these data, the structure of compound 1 was determined as 19-O-β-D-glucopyranosyl-labda-8(17),14-dien-13-ol.

Key HMBC and ROESY correlations of compound 1.
By comparison of the NMR data with the literature, nine known compounds were identified as 23-hydroxy-3-oxoolean-12-en-28-oic acid (2) [12], 3,23-hydroxy-olean-12-en-28-oic acid (3) [13], α-boswellic acid (4) [14], wilforlide B (5) [15], wilforlide A (6) [15], olean-12-en-28-oic acid (7) [16], 3α,28-dihydroxy-olean-12-en-29-oic acid (8) [6], 3-oxo-21-hydroxy-olean-12-ene (9) [17], and oleanolic acid (10) [18].
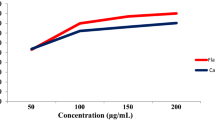
A target assay based on effects against superoxide anion generation and elastase release by human neutrophils in response to fMLP/CB was carried out for compounds 1–10. The results showed that compounds 2 and 3 indicated anti-inflammatory activity against superoxide anion generation and elastase release (IC50 = 1.53 and 1.93 μM for 2; IC50 = 4.39 and 1.92 μM for 3).
Experimental
Genaral. Column chromatography (CC): silica gel (200–300 or 100–200 mesh, Qingdao Marine Chemical Factory, P. R. China), MCI gel (75–150 μm, Mitsubishi, Japan), Lichroprep RP-18 (40–63 μm, Merk, Germany) and Sephadex LH-20 (Amersham Biosciences AB, Uppsala, Sweden). Thin-layer chromatography (TLC): silical gel GF254 plates (Qingdao Marine Chemical Factory, P. R. China), visualization by spraying with 10% H2SO4–EtOH, followed by heating on a hot plate. Semipreparative HPLC: Agilent 1200 system with a Zorbax SB-C18 column (5 μm, 9.4 × 250 mm). Optical rotations: Jasco DIP-370 digital polarimeter. 1D and 2D NMR Spectra: Bruker AM-400, DRX-500, and Avance III 600 spectrometers with TMS as the internal standard. EI-MS and HR-EI-MS Spectra: VG Auto Spec-3000 spectrometer.
Plant Material. The aerial parts of T. hypoglaucum were collected in Cang Mountain, Dali, Yunnan Province, China, in September 2011. The sample was identified by Dr. Yong-Peng Ma, Kunming Institute of Botany, Chinese Academy of Science, and a voucher specimen (KMUST 2011092601) has been deposited in our laboratory.
Extraction and Isolation. Air-dried and powdered aerial parts of T. hypoglaucum (10 kg) were extracted with 80% acetone (3 × 15 L, 1 d, each) at room temperature and then concentrated in vacuo to give an extract, which was suspended in H2O and then successively extracted with EtOAc (4 × 4 L) and n-BuOH (3 × 4 L). The EtOAc extract (460 g) was subjected to silica gel CC eluting with CHCl3–acetone (10:0, 9:1, 8:2, 7:3, 6:4, v/v) to give five fractions (Frs. A–E). Fraction B (21.5 g) was chromatographed on a MCI column (MeOH–H2O, 90% and 100%) to obtain five subfractions (Subfrs. B1–B5). Subfraction B1 (1.11 g) was subjected to silica gel column chromatography using petroleum ether–acetic ester to give 1 (12.0 mg). Subfraction B2 (5.5 g) was applied on a Lichroprep RP-18 gel column, eluting with MeOH–H2O (50, 70, 90 and 100%) to give four subfractions (Subfrs. B2.1–B2.4). Subfraction B2.2 (1.8 g) was subjected to silica gel column chromatography using CHCl3–MeOH (300:1) and purified by Sephadex LH-20 column to obtain compound 2 (5.0 mg). Fraction C (19 g) was chromatographed on a Sephadex LH-20 column (60 × 600 mm), using MeOH–H2O (30, 60, 90, and 100%) as eluent to give five subfractions (Subfrs. C1–C5). Subfraction C4 (2.4 g) was subjected to silica gel CC with CHCl3–MeOH (1:0, 100:1, 25:1, 10:1, 1:1, 0:1) to afford six subfractions (Subfrs. C4.1–C4.6). Subfraction C4.1 (1.5 g) was subjected to silica gel CC, eluting with CHCl3–MeOH (1:0–0:1), to yield subfractions C4.1.1–C4.1.5. Subfraction C4.1.4 (135.0 mg) was applied on a MPLC RP-18 (3 mL/min, MeOH–H2O, 60, 80%) to give compounds 6 (10.0 mg), 7 (5.8 mg), and 8 (6.0 mg). Subfraction C4.2 (1.12 g) was subjected to silica gel CC with petroleum ether–acetone (4:1), then purified by another silica gel column and eluted using petroleum ether–EtOAc (8:1) to yield compounds 5 (7.0 mg) and 10 (15.0 mg). Subfraction C4.3 (0.87 g) was applied on a MPLC RP-18 column (3 mL/min, MeOH–H2O, 65%) to give four subfractions (Subfr. C4.3.1–C4.3.4). Subfraction C4.3.1 (338 mg) was separated on a silica gel column, eluting with petroleum ether–acetone (6:1), to give compounds 3 (5.0 mg), 4 (8.0 mg), and 9 (10.0 mg).
Acid Hydrolysis of 1. Compound 1 (2.0 mg) was hydrolyzed with 1 M HCl (0.5 mL) in a screw-capped vial for 3 h at 90°C. The mixture was evaporated to dryness under vacuum, and then the residue was dissolved in H2O and extracted with CHCl3. The aqueous layer was collected and neutralized by addition of Amberlite IRA400 (OH-form) and filtered. The filtrate was dried in vacuo, then dissolved in 0.2 mL of pyridine containing L-cysteine methyl ester (10 mg/mL) and reacted at 60°C for 1 h. To this mixture a solution (0.2 mL) of trimethylsilylimidazole in pyridine (10 mg/mL) was added, and it was heated at 60°C for 1 h [19]. The final mixture was directly analyzed by GC [30QC2/AC-5 quartz capillary column (30 m × 0.32 mm) under the following conditions: column temperature: 180°C/280°C; programmed increase 3°C/min; carrier gas: N2 (1 mL/min); injection and detector temperature: 250°C; injection volume: 4 μL; split ratio: 1:50]. The standard D-glucose was prepared following the same procedure. Under these conditions, the retention time for D-glucose was 18.29 min; the hydrolysate had the same retention time with it.
The preparation of human neutrophils and measurement of superoxide anion generation and elastase release were carried out following the method in the literature [20].
References
Editorial Board of Flora of China, Flora of China (2005 Ed.), Vol. 45 (3), Science Press, Beijing, 2005, p. 178.
A. M. Brinker, J. Ma, P. E. Lipsky, and I. Raskin, Phytochemistry, 68, 732 (2007).
J. Ma, M. Dey, H. Yang, A. Poulev, R. Pouleva, R. Dorn, P. E. Lipsky, E. J. Kennelly, and I. Raskin, Phytochemistry, 68, 1172 (2007).
H. Q. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. F. Jia, and D. Li, J. Nat. Prod., 62, 1522 (1999).
H. Q. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. F. Jia, and D. Li, Phytochemistry, 53, 805 (2000).
H. Q. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, M. Tori, S. Takaoka, Y. Jia, and D. Li, Tetrahedron, 57, 8413 (2001).
H. Q. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, M. Tori, S. Takaoka, Y. Jia, and D. Li, J. Nat. Prod., 64, 582 (2001).
J. Bao and S. M. Dai, Rheumatol. Int., 31, 1123 (2011).
H. Z. Li, L. Z. Fu, H. M. Li, R. T. Li, and X. L. Deng, Phytochem. Lett., 5, 572 (2012).
A. Ulubelen, G. Topcu, C. Eris, U. Soenmez, M. Kartal, S. Kurucu, and C. Bozok-Johansson, Phytochemistry, 36, 971 (1994).
J. Hu, X. Q. Chen, and Q. S. Zhao, Helv. Chim. Acta, 94, 1085 (2011).
J. J. Li, J. Yang, F. Lu, Y. T. Qi, Y. Q. Liu, Y. Sun, and Q. Wang, Chin. J. Nat. Med., 10, 279 (2012).
H. Kizu and T. Tomimori, Chem. Pharm. Bull., 30, 3340 (1982).
J. Y. Zhou and R. Cui, Acta Pharm. Sin., 37, 633 (2002).
G. W. Qin, X. M. Yang, W. H. Gu, B. D. Wang, Z. X. Chen, R. X. Guo, and K. W. Shao, Huaxue Xuebao, 40, 637 (1982).
C. Djerassi, J. A. Henry, A. Lemin, T. Rios, and G. H. Thomas, J. Am. Chem. Soc., 78, 3783 (1956).
D. Caceres-Castillo, G. J. Mena-Rejon, R. Cedillo-Rivera, and L. Quijano, Phytochemistry, 69, 1057 (2008).
X. L. Fu, L. W. Zhang, W. H. Lin, and Q. S. Li, Zhongcaoyao, 41, 704 (2010).
S. Hara, H. Okabe, and K. Mihashi, Chem. Pharm. Bull., 35, 501 (1987).
S. C. Yang, P. J. Chung, C. M. Ho, C. Y. Kuo, M. F. Hung, Y. T. Huang, W. Y. Chang, Y. W. Chang, K. H. Chan, and T. L. Hwang, J. Immunol., 190, 6511 (2013).
Acknowledgment
Financial support from the National Natural Science Foundation of China (Nos. 21262021 and 31660102) is acknowledged. We also wish to thank Prof. Tsong-long Hwang (Graduate Institute of Natural Products, College of Medicine, Chang Gung University) for his help in the biological assays.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2018, pp. 400–402.
Rights and permissions
About this article

Cite this article
Zhao, Q., Li, HM., Chen, XQ. et al. Terpenoids from Tripterygium hypoglaucum and Their Anti-Inflammatory Activity. Chem Nat Compd 54, 471–474 (2018). https://doi.org/10.1007/s10600-018-2381-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-018-2381-4