
Abstract
Cancer metastasis follows a sequential series of events, and many of the critical steps are distinctly similar to EMT-like transformations that occur during normal embryonic development. A current area of focus is the similarities between how cancer cells interact with the ectopic parenchyma after metastatic spread, and secondary developmental MET events that occur in epithelial tissues that have re-assembled within the embryo from mesenchymal cells. Accumulating evidence suggests a critical role for these secondary events, termed mesenchymal-epithelial transitions (MET) in development and mesenchymal-epithelial reverting transitions (MErT) in cancer. In this situation, metastatic seed cancer cells may inertly become part of the ectopic tissue and therefore surmount the metastatic inefficiencies to which most disseminated cancer cells succumb. Just as a critical EMT event is the downregulation or silencing of E-cadherin, we discuss the role of E-cadherin in cancer-associated MErT at distant metastatic sites and speculate on the implications for the fate of micrometastases that undergo a transition to being E-cadherin positive.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Carcinoma dissemination follows from an epithelial-to-mesenchymal-like transition (EMT)
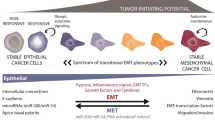
The major part of cancer morbidity and mortality occurs upon the dissemination of carcinomas from their original sites of development. While any tissue can undergo a neoplastic transformation, the majority of tumors derive from the epithelial cells of tissues. These carcinomas are distinguished from their normal counterparts by a loss of tissue and cell organization (Fig. 1) [23].

Carcinomagenesis and progression entails multiple phenotypic changes. Epithelial tissues consist of sheets of normal cells (stippled black) linked together by homotypic binding of E-cadherin (thick black bars). This establishes a polarity that segregates apically secreted factors, such as EGF, from their basolaterally-presented receptors that normally are utilized by stromally-derived factors, such as TGFα. Due to genetic and epigenetic events, the E-cadherins are lost during carcinoma development (lined cells), allowing for autocrine signaling. This ‘dedifferentiation’ is the carcinoma-associated EMT. However, we propose that during metastatic seeding to other epithelial organs E-cadherin is re-expressed enabling linkages to, and signaling from normal parenchymal cells (gray cells). This is concurrent with downregulation of signaling from the previous autocrine factor/receptor loops. This characterizes the mesenchymal-to-epithelial reverting transition (MErT). Adapted from Kim et al. [23]
In the normal situation, the epithelial cells are in directed communication with the stromal compartment via an organized structure. Epithelial cells are arranged mainly in sheets (with the exception of skin) with adjacent cells tightly connected via tight junctions and gap junctions. These connections not only serve for communication and limit both cell proliferation and migration, but also establish a cell polarity. At the apical surface in most tissues, the epithelial cells are in contact with body fluids that are either wholly or partly produced by these same cells. These fluids contain biologically factors, growth factors in particular, that are usually inert to the tissue as the receptors for such factors are sequestered on the baso-lateral surfaces. The tight junctions, constructed upon homotypic binding of Epithelial-Cadherins (E-cadherin or CDH1), limit the access of the apical fluids to the basolateral spaces and surfaces and the underlying stromal compartment.
Upon neoplastic transformation, this orderly arrangement is lost. A host of genetic and epigenetic changes occur that lead to loosening and even loss of the tight junctions (for reviews see: [4, 13, 16]). The consequences of this are manifold. First, the epithelial cells are now exposed to potential autocrine factors, as the localization of ligand production and receptor expression are no longer segregated. Second, the epithelial cells are released from physical restraints with loss of contact inhibition, allowing from cell movement and even proliferation. And third, the stromal elements are now exposed to many epithelial-derived bioactive factors promoting a stromal response. This latter event may change the profile of signals that derive from the stromal compartment, as the composition and differentiation state of the stromal elements change. The net sum of these tissue alterations are that the epithelial cells are directed to assume a less differentiated state that converges towards the mesenchymal phenotype characterized by motility and proliferation with a more fluid cell architecture that has limited cell–cell direct communications. This is the so-called epithelial-to-mesenchymal-like transition (EMT) of carcinomas [41, 42].
This carcinoma EMT is distinguished from true EMT situations noted in development by the fact that the cells do not actually subsume a full or physiological role similar to a mesenchymal-derived (usually stromal/fibroblastoid) cell [4, 16, 41, 42]. Rather the attributes of the cells converge on those noted in stromal cells, and the normal epithelial markers and functions are lost. This situation is not unique to carcinoma de-differentiation; incomplete EMT is also noted during wound healing. During skin repair, for example, the basal keratinocytes bordering the missing tissue downregulate their defining cytokeratin and E-cadherin structures and change their complement of adhesion receptors to assume a mesenchymal-like phenotype that is proliferative, migratory and synthetic for dermal matrix components [1, 20]. Interestingly, these same cells then ‘revert’ or redifferentiate into basal keratinocytes when the wound defect is re-epithelialized. Thus, the first steps in the dedifferentiation process of carcinoma progression are an EMT process not dissimilar from the post-natal wound-related EMT.
Despite the long-standing recognition of this carcinoma EMT and recent findings as to underlying molecular controls (though these are not specific for EMT) [29], there is no accepted molecular definition of what constitutes this process [41, 42]. This may be due to the multitude of pathways carcinomas may take to achieve this aggressive phenotype. A picture is emerging of common molecular findings that account for both the histopathological picture and the cell biology findings. At the minimum, the carcinoma EMT is defined by a loss of normal epithelial architecture, namely both the physiological homogenous asymmetry of the cell and the cell–cell contacts and communications that anchor this. At the molecular level, this is reflected by downregulation or loss of specific cytokeratins and E-cadherin. The latter is of particular importance, because it can be viewed as both a cause and effect of the EMT.
In addition to loss of E-cadherin functionality, the structural proteins in these tumor cells undergo a change, with downregulation or loss of cytokeratins and emergence of the mesenchymal marker vimentin [4, 13, 16]. Often, N-cadherin upregulation is coincident with EMT and contributes to tumor progression [5]. However, this adhesion/attachment molecule is not a consistent marker for EMT, also appearing on cells along side E-cadherin. Another cell surface marker that is often linked to the EMT is ZO-1 [19], though again, this is an inconsistent marker with a more quantitative than qualitative change. A number of differentiation-related transcription factors contribute to this proteome change, though these are specific neither to EMT nor to mesenchymal cells [22, 29]. As such, in the absence of truly specific markers for either the epitheloid or mesenchymal phenotype, the definition of EMT is one of overall interpretation of cell phenotype. However, for carcinoma cells the most consistent molecular marker is E-cadherin, and our discussion will focus on the expression and functionality of this, though studies should examine additional molecular and phenotypic/morphometric markers when querying EMT.
Loss of E-cadherin disrupts not only cell–cell junctions but also allows for loss of the normal organ architecture. The cells are now free to move both horizontally and vertically within the epithelial layer as they are no longer constrained within a functional syncytium [3, 10] (Fig. 1). This is a histopathological hallmark of neoplastic transformation. Furthermore, the absence of apical-basal barriers between the cells enables factors produced by the cells to reach cognate receptors normally segregated on the basolateral surfaces. This autocrine signaling often further reinforces the EMT phenotype as stimulation of most receptors with intrinsic tyrosine kinase activity, particularly the ubiquitous EGF receptor system, drive E-cadherin downregulation and epithelial dedifferentiation in themselves [24]. Lastly, the E-cadherin-based cell connection can control global cell signaling, as the catenins that anchor these plaques possess location-dependent signaling properties [14, 30] (Fig. 2). At one, albeit simplistic level, E-cadherin-based plaques can be viewed as sequestering β-, γ- and p120-catenins for an epithelial signaling mode and preventing their ‘transformative’ effects on the cell transcriptome. The extent of the global control on cell phenotype exerted by E-cadherin expression can be seen by its designation as a tumor suppressor; forced re-expression of E-cadherin in negative carcinoma cells can revert the neoplastic phenotype [38]. For these reasons, E-cadherin expression on the cell surface has emerged as a molecular hallmark of the carcinoma EMT, and will be the focus on this minireview.

E-cadherin sequesters catenins and controls their signaling in addition to forming cell–cell adhesions. (a) E-cadherin sequesters β- and p120-catenins on its intracellular catenin binding domains. In an untransformed cell, p120 is thought to stabilize E-cadherin at the surface, β-catenin is sequestered from forming a complex with axin and, in this location also functions as an adaptor protein for a-catenin, which in turn anchors E-cadherin to the cytoskeleton. (b) In many carcinomas, E-cadherin is silenced by promoter methylation allowing β-catenin to translocate to the nucleus and p120 to promote an epithelial phenotype. However the mechanisms of how these catenins act and wether they act individually or in concert are not settled. (c) E-cadherin dependent adhesion in itself is not a dominant stop mechanism to inhibit invasion. In studies where the β-catenin binding domain was deleted from the E-cadherin intracellular domain, but E-cadherin was still able to mediate adhesion through direct crosslinking with α-catenin and therefore interaction with the actin cytoskeleton, β-catenin was free to signal in the cell cytoplasm and led to an invasive phenotype (though this phenotype was independent of its TCF-mediated transcriptional activity). Therefore, cytoplasmic localization of β-catenin is thought to contribute to the mesenchymal nature of cells. (d) In studies of cell that had low levels of E-cadherin and cytoplasmic localization of p120, tyrosine phosphorylation on p120’s amino-terminal by the pro-oncogene Src was thought to contribute to modulate its contribution to cell migration. When p120 is knocked down, the equilibrium shift to Rho-GDP promotes actin polymerization, stress fiber formation, a flattened morphology and less invasive phenotype. Therefore, cytoplasmic localization of p120 is thought to contribute to the mesenchymal nature of cells. These four scenarios provide data that show the critical role of E-cadherin as a signal modulation molecule by sequestering catenins, primarily p120 and β-catenin. In the absence of E-cadherin homotypic binding, this plaque is unstable and the catenins are now free to relocalize
Metastatic seeding is rate-limiting for carcinoma progression
Key to carcinoma pathology is the ability of carcinoma cells to survive and thrive in ectopic tissues after their escape from the primary tumor mass (Fig. 3). As noted above, the carcinoma EMT is critical to this initial escape by enabling individual cell migration and invasion through barrier matrices [43]. In order to establish metastatic foci, some of these same properties come into play [26]. First, the cells arrest in the dissemination conduits (lymph and blood vessels) by size mismatch of the tumor cells being larger than the bore of capillaries. The carcinoma cells then must penetrate between the endothelial cells and migrate into the ectopic tissue. This appears to be relatively efficient. However, the main rate-limiting step appears to be the ability of these cells to ‘set up house’ in this foreign milieu.

Carcinomagenesis and metastatic dissemination requires diverse behaviors and phenotypes at different stages. Initially, the primary tumor cells need to lose their cell–cell adhesion (mainly E-cadherin mediated) to break from the tumor mass and enter vascular conduits via active migration. Survival in the vascaulature through at least one capillary bed requires distinct behaviors of architectural malleability and resistance to anoikis. Arrest at the target organ is usually via physical arrest, but the tumor cells must then recognize the endothelial lining and transit this barrier. We propose that establishment and longer term survival entails re-expression of E-cadherin along with some redifferentiation at least initially. It is possible and even likely that when the micrometastasis progress to clinically-relevant lesions and even seed other sites, that this transient MErT itself reverts as the metastatic lesion becomes more aggressive with autocrine signaling reminiscent of the primary aggressive lesion
Many years ago it was appreciated that tumor cells disseminate preferentially to ectopic tissues based mainly on the ability of that host tissue to support the survival and proliferation of those cells [9]. This is referred to as the ‘seed and soil’ hypothesis, first conceptualized over a century ago [31]. The stromal component of different tissues and organs produce and present overlapping but distinct matrix components and secreted factors that serve to ‘feed’ the local cells. Disseminated tumor cells must find a host environment that provides the necessary signals or these cells must provide the missing factors in an autocrine or induced manner. Some of these missing factors might be replaced by genetic mutations that alleviate a particular requirement or are provided by the autocrine signaling inherent in the carcinoma EMT. Still these carcinoma-inherent signals do not allow for micro-environment-independent proliferation. As an example, aggressive metastatic prostate carcinoma cell lines still present a preference for growth in the presence of bone marrow- and liver-derived fibroblasts [11], the two major sites of prostate carcinoma metastasis [8, 36].
The survival of these carcinoma cells in ectopic tissue is complicated by the question of tumor cell dormancy. While tumor masses need to approach a billion cells (or less than 30 doublings) to be detectable as metastases, this should occur within months if the tumor cells continue to proliferate at the inherent cell-doubling rate quantifiable in most highly aggressive primary tumors. However, in many tumors, metastases appear only years after removal of the primary mass. During this time, the metastatic foci must remain minimal, being either in a state of balanced proliferation-apoptosis/death or as a dormant carcinoma cell. The key difference between these two options is that in the former, the local milieu must present a full complement of factors for cell proliferation plus signals for death, whereas in the latter, only a partial complement of factors needs to be present to enable cell survival at a lower metabolic load.
The question of ectopic site preference, survival, and proliferation reduces to that of metastasis micro-environmental milieu factors. While there are nascent efforts to fully catalogue tumor microenvironments components, some hints can be gleaned from the carcinoma cells themselves. What has been surprising is that many metastatic carcinomas appear less dedifferentiated than the primary tumor from which they derive [25, 45]. This determination is based on morphometric criteria upon pathological examination, buttressed by molecular findings in these biopsies. In fact, it is well established that many breast cancer metastases to the liver seem to recreate hepatocyte cords with carcinoma cells [39] (Fig. 4). Liver carcinomae have long been known to form glandular elements in liver metastases even when the primary lesion appears quite dedifferentiated; though, only recently have attempts to demonstrate reversion of the EMT been put on a molecular basis [19]. Further, we have found that in at least some prostate cancer metastases to the liver, the E-cadherin adhesion complex is re-expressed co-incident with cytokeratin and in the absence of vimentin and other markers of progression in the primary prostate carcinoma site [45]. This is counter-intuitive to the prevailing view that carcinomas continue down a dedifferentiation EMT cascade as they get more aggressive and disseminate. Rather these reports suggest that the actual metastatic foci may be more epitheloid than the primary site.

Breast cancer metastases to the liver can subsume a hepatocyte morphology. Not the organ morphology flows seamlessly from the liver cords (right) through the breast cancer metastases (left). From Stessels et al. [39]
Examining these metastases with immunohistochemistry confirmed this more differentiated phenotypes in many carcinomas. Interestingly, a number of reports related E-cadherin membrane staining in the metastases but not in the primary carcinoma sites [17, 25, 32, 35]. We have found that prostate carcinoma metastases to the liver present a differentiated profile with cytokeratins and E-cadherin being present and vimentin absent with downregulation of EGF receptor signaling [46]. Thus, at first blush, it appears as if the metastasis did not undergo the carcinoma-related EMT.
A mesenchymal-to-epithelial reverting transition (MErT) occurs during metastatic seeding
The finding of membrane-expressed E-cadherin in carcinoma metastases leaves us with two possibilities. The first is that the metastases formed from E-cadherin positive carcinoma cells that escaped the primary mass. Even though many of these primaries were ‘scored’ as E-cadherin-negative (or severely downregulated), this possibility cannot be excluded as the larger primary tumors examined show heterogeneity of expression with some nests remaining more differentiated. Still, the clinicopathological correlation between low to absent E-cadherin and tumor dissemination is strong [24], and the cellular mechanisms undergirding this model are logical and repeatedly demonstrated. Carcinoma cells that present surface E-cadherin are limited as to migration and invasiveness in vitro, and do not form distant metastatic tumors in animal models. However, given the rare nature of metastases despite relatively frequent shedding of cells from the primary mass [7], these metastases could arise from the unusual escape of an E-cadherin-positive cell in the primary mass, if E-cadherin expression provided a survival and proliferation advantage (see below). This question of dissemination, therefore, cannot be addressed by human tumor surveys, but rather requires experimental probing using clonal carcinoma cell populations. In what appears to be the first report directly addressing this question, we have found that when the highly aggressive and E-cadherin-negative MDA-MB-231 breast carcinoma cell line forms lung metastases from orthotropic primary tumors in the inguinal mammary fat pads of mice, these metastases express E-cadherin [37]. While this is far from conclusive, it does present a proof a principle that E-cadherin-expressing metastases might arise from E-cadherin-negative cells.
Re-expression of E-cadherin in the metastatic site, therefore, represents the second possibility (Figs. 1 and 3). While this is consistent with the histopathological correlations, E-cadherin re-expression would represent a tumor cell plasticity, that of reverting the carcinoma-associated EMT, that has not been demonstrated to-date. The nature of E-cadherin downregulation mechanisms suggests how this could be accomplished.
E-cadherin surface expression is downregulated by two known mechanisms, functioning separately at the post-translational and the transcriptional levels (Fig. 5). Tyrosine kinase signaling, noted both in response to various growth factors and during neoplastic progression, can downregulate E-cadherin secondary to phosphorylations of the E-cadherin complex [15, 34]. Scatter factors (such as HGF and EGF) are so named as they lead to epithelial cell migration away from cohesive masses secondary to E-cadherin downregulation [2, 3, 28]. The dissociation of the cytoplasmic aspect of the E-cadherin plaque results in E-cadherin internalization and degradation. This may function in carcinomas, almost all of which have autocrine growth factor signaling loops most often that via the EGF receptor [43]. We have found in prostate carcinoma lines that inhibition of this autocrine EGF receptor loop (and likely the EGFR-induced HGF/c-met autocrine loop [27]), either by direct disruption of the signaling loop or by second site signaling trans-attenuation, results in E-cadherin re-expression and cell–cell cohesion [46, 47]. Thus, post-translational E-cadherin downregulation represents an available target for counter-regulation by other factors that might be present in the metastatic microenvironment.

E-cadherin expression is regulated on multiple levels. The plastic nature of these controls allows for EMT defined and enabled by loss of E-cadherin and MErT when E-cadherin is re-expressed. Methylation is reported to be responsible for E-cadherin silencing in the majority of aggressive and metastatic carcinomas. These epigenetic changes are at the base of the pyramid, as access to the gene in the first place is necessary for its ultimate expression. On the transcriptional level, overexpression of SLUG/SNAIL is responsible for for transcription downregulation of E-cadherin, and is noted in a large subset of cancers. On the effector level, receptor tyrosine kinase phosphorylation causes internalization of E-cadherin from the cell-surface by phosphorylation of E-cadherin effector catenins and modulation of the ubiquitinase Hakai, resulting in either trafficking of E-cadherin for endosomal recycling or degredation. As all of these mechanisms are prevalent in invasive cancers, the pathway to MErT may be diverse as each suppression mechanism is controlled individually
In most other carcinomas, distinct from prostate carcinomas, E-cadherin appears to be shut off at the translational level by promoter hypermethylation [18, 40]. This mode of generating a null phenotype differs from other tumor suppressors that are usually negated by deletions or mutations of the coding DNA. While these latter mechanisms are irreversible by their nature, promoter hypermethylation is readily reversible if only by proliferation-linked failure to maintain methylation. One distinction between the normally irreversible tumor suppressors from E-cadherin is that loss of the former often occurs early in neoplastic transformation whereas E-cadherin is related to tumor progression to dissemination. Still, this mode of transcriptional shutoff is reversible. In fact, it has been noted that E-cadherin promoter methylation is unstable [12]. Recently, we have found that E-cadherin promoter methylation can be selectively loss in breast carcinoma cells when proliferating in the presence of normal hepatocytes [37]. Thus, there exist signals in microenvironment of metastasis target organs that may undo both mechanisms of E-cadherin downregulation.
All the above beg the question of why metastatic carcinoma cells would express E-cadherin; basically, what is the selective advantage. The short answer is that this unknown is the subject of nascent investigations. However, we can speculate that this provides a survival advantage for these cells in a challenging ectopic environment. E-cadherin binding not only constructs a barrier between cells but also initiates intracellular signaling in the cognate cells. Two key pathways activated upon E-cadherin binding are the survival-associated ERK MAP kinase and Akt/PKB cascades [6, 33]. Building upon the seed and soil hypothesis, this E-cadherin binding would provide some signals lost when the carcinoma cells dissociate from their original orthotropic ‘home’. Initially at the start of metastatic seeding, this requires cell-heterotypic E-cadherin binding, which has not been directly reported; later as the metastatic cell proliferates, this E-cadherin binding may be between the carcinoma cells. To support the possibility of carcinoma-parenchyma binding via E-cadherins, histopathological analyses of many tumors suggest close associations between metastatic carcinoma cells and the neighboring parenchyma cells [17, 25, 32, 35]. Furthermore in an ex vivo model of carcinoma metastasis to the liver, we find ultrastructural evidence for close connections [44]. Lastly, we have found that when prostate carcinoma cells are induced to re-express E-cadherin, both DU-145 and PC3 cells can form heterotypic adhesions with rat hepatocytes [46]. These data provide proof of principle that carcinoma cells may re-express E-cadherin in response to the ectopic organ microenvironment so as to establish connections with the resident, non-neoplastic epithelial cells.
A model for carcinoma mesenchymal-to-epithelial reverting transition
The preceding provides for a model of tumor cell plasticity that results in differentiation modulation with E-cadherin playing a cell role in the phenotypic alterations noted in both the primary and metastatic sites. In brief, at the primary site, ill-defined genetic and epigenetic changes lead to E-cadherin downregulation, via receptor tyrosine kinase signaling and/or, more often, promoter hypermethylation. This loss of junctional barriers in a subset of carcinoma cells provides a selective advantage likely due to both the physical freedom to move and reorganize and the biological signals derived from autocrine signaling. It is unknown whether the loss of the epithelial barrier further alters the cancer field stroma in a manner that furthers this carcinoma-related Epithelial-to-Mesenchymal-like Transition. However, the loss of E-cadherin comes at the cost of lacking survival signals transmitted via E-cadherin linkages. This appears not to be limiting as the orthotropic site presents the appropriate panoply of factors and signals to support carcinoma proliferation even in the absence of E-cadherin.
Once free of the physical tethering and cell architectural restraints imposed by E-cadherin binding, the mesenchymal-like carcinoma cells can actively migrate through barriers and enter vascular conduits for dissemination to distant sites. While the cell faces numerous challenges during this dissemination process, the major impediment to metastatic growth appears to be the survival and growth (albeit often delayed or slow) in the ectopic site. This is likely due to the incomplete match of factors and signals in different tissues. Interestingly, it appears that many metastatic carcinoma foci do express E-cadherin, whose downregulation is considered critical if not required for dissemination. This suggests that a very minor subset of the disseminated carcinoma cells receive signals from the metastatic target organ to revert the dedifferentiation process at least partly to enable E-cadherin re-expression. As E-cadherin binding elicits canonical survival signals and prevents anoikis [6, 21, 33], even when the binding is cell heterotypal [37], the selective advantage is obvious. It is this process that we propose as the carcinoma-associated mesenchymal-to-epithelial reverting transition or MErT.
Further investigations are required to establish this reverting transition that we have shown can occur in ex vivo and in vitro model systems, as important for de novo disseminating cancers. These would include interventions that would prevent E-cadherin re-expression as limiting metastatic seeding and those that promote E-cadherin re-expression as increasing metastatic efficiency.
Still, the advantages of E-cadherin re-expression and seeming redifferentiation must be explored. The reversion of the general carcinoma cell phenotype to a less aggressive, less proliferative cell seems counterintuitive particularly in regards to the picture that emerges at the primary site. While activation of ERK MAP kinase and Akt/PKB signaling pathways are positives for survival, these pathways are even more strongly elicited by other factors and external signals, without the suppression of proliferation that ensues from E-cadherin adhesions [38]. Answers are not present, but one can speculate that the combination of survival signals with limited proliferation and lowered metabolic requirements are precisely the keys to metastatic seeding. By and large, fast growing cells are most susceptible to death signals whether due to DNA damage, limited nutrients, absent pro-survival signals, or apoptotic signals. This is well recognized in cancer, as the slowest growing tumors generally are the most resistant to standard chemotherapies. In the ectopic metastatic environment, tumor cells not only carry damaged DNA but face an absence of external factors as reflected in the seed and soil hypothesis [9]. It is also possible that entry of these foreign cells will elicit a localized inflammatory reaction, with the production of prodeath signals. Thus, not only would activation of the prosurvival ERK MAP kinase and Akt/PKB pathways protect the carcinoma cell, but also proliferation and metabolic suppression would provide added survival advantages. This speculation can be tested in models of chemotherapeutic and apoptotic challenge.
Lastly, one must consider the implications of this proposed carcinoma-related MErT. Currently, E-cadherin is categorized as a tumor suppressor due to its protective role against the carcinoma-related EMT at the primary site. However, interventions to prevent E-cadherin downregulation may perversely promote seeding of disseminated cells, as the re-expression phenomenon appears to be highly inefficient in the natural course of cancer. Thus, any E-cadherin-target therapies must be highly cognizant of carcinoma progression dynamics. Second, the formation of cell heterotypic E-cadherin adhesions in the metastatic target organ may result in dormancy at the micrometastasis stage. This would result not only in resistance to chemotherapy with tumor re-emergence despite long courses of chemotherapy, but also have implications for what may lead to delayed relapses. It is tempting to speculate that if the proposed MErT leads to dormancy of early micrometastases, then a secondary insult, independent of carcinoma per se, to the local environment may be what induces renewed carcinoma cell proliferation and escape from E-cadherin-mediated contact inhibition. This, along with other many questions raised by this proposed MErT, awaits further experimentation.
References
Babu M, Wells A (2001) Dermal-epidermal communication in wound healing. Wounds 13:183–189
Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4:915–925
Boccaccio C, Comoglio PM (2006) Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer 6:637–645
Christiansen JJ, Rajasekaran AK (2006) Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 66:8319–8326
Christofori G (2006) New signals from the invasive front. Nature 441:444–450
Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze’ev A (2003) Autoregulation of E-cadherin expression by cadherin–cadherin interactions: the roles of b-catenin signaling, Slug, and MAPK. J Cell Biol 163:847–857
Cristofanilli M, Budd GT et al (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351:781–791
Ewing J (1922) Tumors of the prostate. Neoplastic diseases. WB Saunders Company, Philadelphia, pp 784–785
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3:453–458
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3:362–374
Gleave ME, Hsieh JT, vonEschenbach AC, Chung LWK (1992) Prostate and bone fibroblasts induce human prostate cancer growth in vivo: implications for bidirectional tumor-stromal cell interaction in prostate carcinoma growth and metastasis. J Urol 147:1151–1159
Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG (2000) Methylation patterns of the E-cadherin 5’CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 275:2727–2732
Guarino M, Rubino B, Ballabio G (2007) The role of epithelial-mesenchymal transition in cancer pathology. Pathology 39:305–318
Gumbiner BM (2005) Regualtion of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 6:622–634
Hazan RB, Norton L (1998) The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 273:9078–9084
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW (2007) Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol 213:374–383
Imai T, Horiuchi A et al (2004) Elevated expression of E-cadherin and alpha-, beta-, and gamma-catenins in metastatic lesions compared with primary epithelial ovarian carcinomas. Hum Pathol 35:1469–1476
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Kaihara T, Kawamata H, Imura J, Fujii S, Kitajima K, Omotehara F, Maeda N, Nakamura T, Fujimori T (2003) Redifferentiation and ZO-1 reexpression in liver-metastatized colorectal cancer: possible association with epidermal growth factor receptor-induced tyrosine phosphorylation of ZO-1. Cancer Sci 94:166–172
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784
Kang H-G, Jenabi JM et al (2007) E-cadherin cell–cell adhesion in Ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res 67:3094–3105
Kang Y, Massague J (2004) Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118:277–279.
Kim H, Turner T, Kassis J, Souto J, Wells A (1999) EGF receptor signaling in prostate development. Histol Histopathol 14:1175–1182
Kopstein L, Christofori G (2006) Metastasis: cell autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci 63:449–468
Kowalski PJ, Rubin MA, Kleer CG (2003) E-cadherin expression in primary carcinoma of the breast and its distant metastases. Breast Cancer Res 5:R217–R222
Luzzi KJ, MacDonald IC et al (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153:865–873
Mamoune A, Kassis J et al (2004) DU145 human prostate carcinoma invasiveness is modulated by urokinase receptor (uPAR) downstream of epidermal growth factor receptor (EGFR) signaling. Exp Cell Res 299:91–100
Miura H, Nishimura K et al (2001) Effects of hepatocyte growth factor on E-cadherin-mediated cell–cell adhesion in DU145 prostate cancer cells. Urology 58:1064–1069
Moustakas A, Heldin C-H (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 98:1512–1520
Nelson WJ, Nusse R (2004) Convergence of wnt, b-catenin, and cadherin pathways. Science 303:1483–1487
Paget S (1989) The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 8:98–101
Putz E, Witter K et al (1999) Phenotypic characteristics of cell lines derived from disseminated cancer cells in bone marrow of patients with solid epithelial tumors: establishment of working models for human micrometastases. Cancer Res 59:241–248
Reddy P, Liu L et al (2005) Formation of E-cadherin mediated cell–cell adhesion activates Akt and mitogen activated protein kinase (MAPK) via phosphatidylinositol 3 kinase and ligand-independent action of epidermal growth factor (EGF) receptor in ovarian cancer cells. Mol Endocrinol 19:2564–2578
Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z (1994) Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol 14:8333–8342
Rubin MA, Mucci NR, Figurski J, Fecko A, Pienta KJ, Day ML (2001) E-cadherin expression in prostate cancer: a broad survey using high-density tissue microarray technology. Hum Pathol 32:690–697
Shah RB, Mehra R et al (2004) Androgen-independent prostate cancer is a heterogeneous group of diseases. Cancer Res 64:9209–9216
Shepard CR, Yates CC, Wells A (2007) Signaling pathway activation upon re-expression of E-cadherin in invasive breast cancer cells and interaction with ectopic normal epithelial cells (unpublished data)
Simcha I, Geiger B, Yehuda-Levenberg S, Salomon D, Ben-Ze’ev A (1996) Suppression of tumorigenicity by plakoglobin: an augmenting effect of N-cadherin. J Cell Biol 133:199–209
Stessels F, VandenEynden G et al (2004) Breast adenocarcinoma liver metastases, in contrast to colorectal liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer 90:1429–1436
Strathdee G (2002) Epigenetic versus genetic alterations in the inactivation of E-cadherin Seminars. Cancer Biol 12:373–379
Tarin D (2005) The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 65:5996–6000
Thompson EW, Newgreen DF (2005) Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition. Cancer Res 65:5991–5995
Wells A (2000) Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res 78:31–101
Yates C, Shepard CR et al (2007a) Novel three-dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res 96:225–246
Yates C, Shepard CR, Stolz DB, Wells A (2007b) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96:1246–1252
Yates CC, Shepard CR, Stolz DB, Wells A (2007c) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96:1246–1252
Yates C, Wells A, Turner T (2005) Luteinizing hormone releasing hormone (LHRH) analog reverses the cell adhesion profile of DU-145 human prostate carcinoma. Br J Cancer 92:366–375
Acknowledgements
The authors thank the support from the VA Merit Program, the DoD’s Congressionally Directed Medical Research Programs on Prostate and Breast Cancer, and the National Institute of General Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wells, A., Yates, C. & Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis 25, 621–628 (2008). https://doi.org/10.1007/s10585-008-9167-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-008-9167-1