
Abstract
Carcinoma cells that are induced to suppress their epithelial features and upregulate mesenchymal gene expression programs acquire traits that promote an invasive and metastatic phenotype. This is achieved through the expression of a program termed the epithelial-to-mesenchymal transition (EMT)—a fundamental cell-biological process that plays key roles in embryogenesis and wound healing. Re-activation of the EMT during cancer promotes disease progression and enhances the metastatic phenotype by bestowing upon previously benign carcinoma cell traits such as migration, invasion, resistance to anoikis, chemoresistance and tumour-initiating potential. Herein, we discuss recent insights into the function of the EMT and cancer cell plasticity during cancer progression, with a focus on their role in promoting successful completion of the later stages of the metastatic cascade.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The acquisition of mesenchymal cell traits by previously epithelial carcinoma cells is associated with the malignant progression of these cells. The program that facilitates these changes is termed the epithelial-to-mesenchymal transition (EMT), which represents a highly coordinated cell-biological program that concomitantly suppresses epithelial cell markers while upregulating mesenchymal ones [1]. The EMT program can be manifested in cells to various degrees, leading to the idea that cells transit through a series of states arrayed along the epithelial-mesenchymal spectrum. This notion underlies the derivation of the term “partial-EMT” state, where epithelial characteristics are retained alongside newly acquired mesenchymal ones. Thus, rather than considering the EMT as a program that clearly demarcates cells residing in either a definitive epithelial or mesenchymal state, the EMT can be portrayed as a plastic program, whereby cells may be coerced to move to various extents toward a mesenchymal state and may, in the reverse direction, return to their epithelial roots, doing so more readily than previously thought. As our understanding of the EMT program continues to broaden, so too does its centrality to cancer progression and metastasis.
A core transcriptional network involving the actions of a small group of master regulators of the EMT program, specifically EMT-inducing transcription factors (EMT-TFs), provides insights into how the EMT program is induced in response to various afferent signals impinging on normal and neoplastic epithelial cells. Slug, Snail, Twist and Zeb1 are prominent EMT-TFs orchestrating the various manifestations of the EMT program [2, 3]. Activation of different combinations of EMT-TFs can confer on carcinoma cells migratory and invasive capacity, which facilitates their movement out of primary tumour sites and into the circulation, thereby enabling the metastatic potential of EMT-responsive cells [4, 5].
In most carcinomas, only a minority of cells exhibit tumour-initiating ability as revealed by implantation of such cells in appropriate mouse hosts. These cells are referred to as tumour-initiating cells (TICs) or cancer stem cells (CSCs). In contrast, the majority of cells within tumours lacks the capacity to seed new foci of the disease at distant anatomical sites [6]. Of direct relevance to EMT and the process of metastatic seeding is the observation that activation of an EMT program can endow carcinoma cells with tumour-initiating potential [7, 8]. Hence, the EMT can equip benign epithelial cancer cells with the traits necessary to travel to a metastatic site and, thereafter, initiate secondary tumour growth. At the same time, carcinoma cells that have undergone the EMT exhibit heightened resistance to various forms of existing anti-tumour therapies, making this program highly relevant to clinical oncology [9–14].
For these reasons, the ability to define the extent of EMT activation within individual cells or within a tumour as a whole becomes a useful indicator of tumour aggressiveness. This highlights the importance of defining the molecular mechanisms underlying control of the EMT, since such understanding may lead to the development of therapies that can modulate carcinoma cell plasticity and even prevent the development of metastatic disease. Herein, we discuss recent advances in the field of EMT research and cancer metastasis and their implications for future studies.
2 Induction of the EMT
The EMT program is activated in epithelial cells in the context of normal embryogenesis and wound healing [15]. In both situations, this program is activated in individual epithelial cells through signals that these cells receive from their neighbours. Similarly, in the context of carcinoma pathogenesis, contextual signals seem to be responsible for the activation of previously latent EMT programs within individual tumour cells. In carcinomas, these contextual signals derive mostly from the recruited cells that form the stroma of these tumours, often termed the tumour microenvironment. Included among these mesenchymal cells are myofibroblasts, mesenchymal stem cells and an array of inflammatory cells that operate as various components of the innate immune response [2, 16, 17].
The influence of the tumour microenvironment on the epithelial cancer cells’ phenotype is mediated via a series of heterotypic cell-cell signalling molecules, among which Wnt, TGFβ and Notch ligands play a central role. As research in the connections between EMT and carcinomas progresses, the number of signalling players implicated in driving the EMT continues to grow. In addition to the contribution of the above-mentioned canonical molecules, a wide range of growth factors have also been implicated in triggering the EMT program, including the epidermal growth factor (EGF), insulin growth factor (IGF), hepatocyte growth factor (HGF), fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF). Also, hypoxia-inducible signals involving the transcription factor HIF1-α, as well as inflammatory signals (NF-κB) and cytokines, such as interleukin-1β (IL-1β) and the tumour necrosis factor alpha (TNFα), have been shown to cooperate in the complex network of signals that determine EMT activation within individual carcinoma cells (reviewed in 2, 14, 18). As is implied by these findings, heterotypic interactions occurring between carcinoma cells and those residing in the tumour microenvironment are the predominant conduit for activation of the EMT program.
During development, the EMT is tightly regulated, giving evidence of the fact that normal epithelial cells establish safeguards that prevent their being overly responsive to signals that would destabilize their residence in the epithelial state. Hence, simply providing cancer cells with cocktails of EMT-inducing signalling proteins does not necessarily result in induction of the EMT in those cells. For example, in the case of breast cancer cells, downregulation of endogenously synthesized inhibitors of TGFβ and Wnt signalling must also be achieved in order for those cells to become responsive to EMT-inducing signalling proteins [17]. Similarly, disruption and shutdown of the endogenous negative feedback loops that operate between microRNAs and EMT-TFs that suppress expression of the latter, e.g. the Snail1-miR34 or ZEB1-miR200 loops, may prove essential in order to achieve activation of an EMT program and maintenance of its expression [19–21].
The induction of an EMT may be governed by yet other mechanisms operating within carcinoma cells. Thus, the responsiveness of breast cancer cells to contextual EMT-inducing signals may vary strongly, depending on the phenotypic state of these cells. For example, we have shown that breast cancer cells of the basal subtype are poised to respond rapidly and efficiently to EMT-inducing signals, notably those conveyed by TGFβ, by converting to a more mesenchymal, tumour-initiating cell state both in vitro and in vivo. In luminal-type breast cancers, however, we found that the epithelial cancer cells were unresponsive to the same EMT-inducing signals and therefore remained locked in an epithelial cell state [22]. Another well-documented example supporting this notion would be the varied cellular responses to TGFβ; in some cases, cells are able to escape the growth-inhibitory effects of TGFβ and continue to proliferate, while in others, they may activate expression of EMT programs [23, 24].
These observations point to the issue of the intrinsic responsiveness of cancer cells to various types of inductive signals and the molecular mechanisms that determine this responsiveness. We have shown that the chromatin configuration within a cancer cell can play an important role in a cancer cell’s potential to activate a latent EMT program in response to appropriate inductive signals. Thus, basal-type epithelial breast cancer cells govern expression of a master regulator of the EMT program—the ZEB1 EMT-TF—in a way fundamentally different from their luminal breast cancer counterparts. In particular, the basal cells maintain the transcriptional promoter of the ZEB1 gene in a bivalent chromatin configuration, in which both inductive and repressive transcriptional signals are carried by the histones associated with this promoter. This epigenetic configuration, involving the H3K4me3 and H3K27me3 modifications, is termed a bivalent or poised regulatory state, which enables the rapid activation of ZEB1. More specifically, this configuration poises these cells to a rapid induction of EMT-associated cell-biological phenotypes in response to a cell’s exposure to TGFβ. In contrast, luminal-type epithelial cancer cells that are unresponsive to EMT-inducing signals display only the repressive H3K27me3 mark at the ZEB1 promoter, which appears to lock this promoter, and thus the ZEB1 gene, in a state that is unresponsive to TGFβ, thereby enabling the luminal cells to continue to dwell in an epithelial state in the presence of significant concentrations of this cytokine [22].
While the chromatin configuration of an epithelial cancer cell might predict whether it will be responsive to EMT-inducing stimuli, another component of the decision-making process is the intracellular signalling configuration of the cell. For example, the varied functions of TGFβ can be attributed to the activation of specific downstream signalling pathways, where canonical TGFβ/Smad signalling drives the EMT in responsive cells. In addition, however, activation of TGFβ/Erk/MAPK signalling in the same epithelial cell promotes maintenance of the epithelial phenotype by preventing further TGFβ/SMAD pathway activation [25].
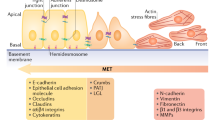
Together, these data demonstrate that successful induction of an EMT requires the appropriate signals and a responsive target cell. This suggests, in turn, the possibility that the future biological behaviour of tumour cells primed to respond to EMT-inducing signals—cells that are therefore likely to form aggressive tumours—might well be predicted at early stages of carcinogenesis, for example, by the configuration of their transcription-regulating chromatin modifications (Fig. 1).

Spectrum of EMT phenotypes in cancer. Some epithelial cancer cells are not responsive to EMT-inducing signals and are unable to undergo the EMT (non-responsive epithelial cancer cell). Yet other epithelial cancer cells are indeed responsive to EMT-inducing signals (responsive epithelial cancer cell). Following disruption of the autocrine signals that previously maintained their epithelial cell state, they transition toward a stable mesenchymal cancer cell state. Once the cancer cells have undergone a complete EMT, autocrine signalling can maintain the resulting mesenchymal phenotype in the absence of the EMT-inducing signals. As epithelial cancer cells move toward that mesenchymal state, they transition through partial-EMT states, thought to be reversible and possibly transient. Progression through the EMT is associated with acquisition of tumour-initiating potential, which peaks at some point along the partial-EMT spectrum; however, further progression into a stable, highly mesenchymal cancer cell state diminishes tumour-initiating potential. Listed below are selections of factors that favour residence in either the epithelial or mesenchymal cell states. (EMT, epithelial-to-mesenchymal transition; EMT-TF, EMT-transcription factors; MET, mesenchymal-to-epithelial transition)
3 Dynamic regulation of the EMT
The studies described above shed little light on another aspect of the EMT and its effects on cells: what determines the stability with which carcinoma cells will reside in one or another phenotypic state, such as the initial epithelial state or the fully mesenchymal state resulting from passage through an entire EMT program? In fact, producing answers to this question is complicated by the fact that carcinoma cells activating an EMT program usually advance only partway toward the mesenchymal state, suggesting the possibility that carcinoma cells might adopt and reside in multiple intermediate phenotypic states lying along the epithelial-mesenchymal spectrum.
Stable or metastable residence in specific phenotypic states might be transmitted through multiple cell generations by cell-heritable chromatin configurations of the type described above. Yet, other types of stably expressed phenotypes might be supported by more complex physiological mechanisms, such as self-reinforcing positive feedback signalling loops created by autocrine EMT-inducing signals. For example, epithelial cells that have advanced significantly toward a mesenchymal state will begin to make and secrete significant amounts of TGFβ, the same protein that previously drove the activation of an EMT program in these cells; once produced, this can impinge on the cells that have just produced them to induce maintenance of EMT program expression, creating a self-reinforcing autocrine signalling loop [17, 26]. Such autocrine signalling may ensure residence in a more mesenchymal state even in the absence of ongoing contextual signals received by these cells from the surrounding tissue microenvironment.
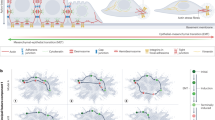
The stability of residence in more mesenchymal states holds implications for the clinical behaviour of these cells. For example, as mentioned earlier, an advance partway through an EMT program is associated with the ability to enter into the tumour-initiating state, often termed the cancer stem cell state. Expression of tumour-initiating ability would seem to be critical to the ability of disseminated carcinoma cells to serve as the founders of new metastatic colonies. This requirement may compromise the success of metastasis formation. Thus, cancer cells leaving the primary tumour will no longer experience EMT-inducing signals that were present within and released by the stromal microenvironment of the tumour. In the absence of such signals, these cells may lapse back into the more epithelial state of their distant ancestors, thereby losing the tumour-initiating ability that would seem critical to the successful founding of a metastatic colony (Fig. 2). This highlights the need of disseminated carcinoma cells to sustain an EMT program in a cell-autonomous manner, such as that made possible by the aforementioned autocrine signalling loops.

Summary of proposed EMT dynamics operating during tumour progression. Epithelial cancer cells responsive to EMT-inducing signals can acquire migratory and invasive capacity, as well as tumour-initiating potential. Together, these traits cooperate to enable a carcinoma cell to transition into one with mesenchymal traits that is capable of disseminating and seeding metastatic deposits. This schematic highlights various spatiotemporal situations in which epithelial cancer cells may be exposed to EMT-inducing signals and the subsequent effects on their metastatic success. Minimal induction of the EMT may not be sufficient to promote metastasis; however, maximum induction of the EMT resulting in stably mesenchymal cancer cells leads to suboptimal outgrowth of metastases. In contrast, induction of a partial-EMT optimizes tumour-initiating potential while still maintaining cell plasticity, i.e. the ability to reverse the EMT process and thus undergo an MET, thereby generating more epithelial progeny, whose presence greatly increases the success of metastatic colony formation. Cancer cells that undergo the EMT at the primary tumour site may receive signals to reinforce their phenotype while in the circulation, e.g. from adhered platelets. At the secondary site, cancer cells may receive MET-inducing signals that promote a transition back to an epithelial phenotype and thereby promote secondary tumour growth. MET at the secondary site may also arise due to the absence of EMT-inducing signals. Alternatively, premature loss of EMT traits at the site of potential metastasis formation may cause cancer cells to lose tumour-initiating potential, thereby aborting the formation of metastatic colonies
In fact, some evidence suggests that cancer cells are exposed to EMT-inducing signals even outside the primary tumour microenvironment. For example, after they have entered into circulation, tumour cells are known to become coated with platelets that act to protect the enveloped cancer cell from shear stress and attacks by natural killer (NK) cells and to promote adhesion to the endothelium—attributes that promote metastatic success [27, 28]. Independent of those functions, platelets are rich sources of growth factors and have been shown to induce or maintain the EMT via the release of TGFβ that accompanies platelet activation and degranulation [29]. In principle, these findings might lead to the conclusion that epithelial cancer cells need not undergo the EMT while still residing in the primary tumour, but instead may do so at later stages of the metastatic cascade, e.g. in the circulation. However, given the extensive evidence that expression of components of an EMT program is required for dissemination from primary tumours, it seems more plausible that these platelet interactions amplify and sustain EMT programs that were initiated within carcinoma cells while they still resided in primary tumours (Fig. 2).
In instances where a tumour cell expressing components of the EMT program arrives at a distant anatomical site such as the lungs to take up residence and initiate a new metastatic deposit, some evidence suggests that successful seeding is facilitated by VEGFR1+ haematopoietic cells and Mac1+−myeloid cells that have established a favourable microenvironment prior to the cancer cells’ arrival [30, 31]. Indeed, the premetastatic niche, which is rich in myeloid cells, haematopoietic progenitor cells and microvesicles, is also endowed with a variety of potential EMT-promoting growth factors including VEGF, MMP2, MMP9, COX2 and WNT5A for example [32–34]. It is early days in our understanding of the determinants governing the development of a premetastatic niche in various tissues; the dependence of this process in the lung on the presence of bone marrow-derived VEGFR1+ myeloid cells may help to explain why the bone is a favoured metastatic site for several carcinomas [35]. It will be interesting to determine in the future if components of the premetastatic niche promote metastasis via induction/maintenance of EMT programs in recently arrived cancer cells.
4 Partial-EMT and phenotypic plasticity
The idea of a “partial-EMT”—in which epithelial carcinoma cells advance toward a partially epithelial/partially mesenchymal phenotypic state—may present a metastatic “sweet spot”, in which mesenchymal traits are initially acquired to enable departure from the primary tumour site, survival in the circulation and arrival at a secondary site; at the same time, residence in this mixed epithelial/mesenchymal state may enable progeny carcinoma cells to re-epithelialize at the metastatic site, which seems to be essential for metastatic colonization and development of macrometastatic outgrowths [36–40]. Thus, disseminated carcinoma cells that have advanced through an entire EMT program, thereby shedding all epithelial traits, have been found to be ineffective in seeding metastatic colonies, possibly because they lose the phenotypic plasticity that is critical to the robust outgrowth of a metastatic colony (Fig. 2) [41–43]. Accordingly, cancer cells that undergo the reverse process of the EMT, that is, the mesenchymal-to-epithelial transition (MET), are more adept at seeding metastases [37, 44] and as such must exhibit a degree of phenotypic plasticity.
This notion of a partial-EMT appears to be a critical element in designing experiments seeking to validate the role of the EMT in carcinoma pathogenesis, as it indicates that cancer cells do not need to reach a purely mesenchymal state in order to benefit from an EMT program. For example, in certain tissue contexts, the Snail1 and Zeb1 EMT-TFs are strong repressors of the epithelial phenotype, but only weak inducers of the mesenchymal phenotype. In contrast, Twist1 is a strong inducer of the mesenchymal phenotype but a weak repressor of the epithelial phenotype [15]. In the PyMT mouse model of breast adenocarcinoma development, Snail1 clearly drives the initial stages of mammary tumour development, and Zeb1 is only strongly activated in the latter stages of tumour progression, while Slug contributes minimally to the EMT phenotype of the carcinoma cells [45]. The implication here is that in order to study the EMT in any given system, it is imperative to elucidate the potential drivers and effectors of the EMT in that particular system. Moreover, these studies reinforce the importance of studying multiple facets of the EMT program in order to gain a true representation of its expression and contribution to the metastatic cascade.
5 Prevalence of the EMT in cancer
Many studies in a variety of cancer types have confirmed that overexpression or knockdown of EMT-TFs enhance or inhibit tumorigenesis and metastasis, respectively [15, 46–48]. However, identifying when and where carcinoma cells undergo the EMT in vivo continues to prove challenging in the hands of many. Thus, in many analyses, it is difficult to resolve the partial EMTs undergone by neoplastic cells from the complete EMTs that are naturally a characteristic of nearby stromal cells. Moreover, EMT programs are plausibly only activated by small subpopulations of the neoplastic cells within islands of these cells, creating a further obstacle to simple resolution. In addition, the often-transient nature of the EMT activation makes the timing of analysis during the course of multistep tumour progression critical. Finally, and of relevance, multiple alternative EMT-TFs can participate in orchestrating an EMT and may act redundantly to do so, complicating the interpretation of results obtained by inactivating the gene encoding one or another EMT-TF in the genome of a carcinoma cell.
Recently, using genetically engineered knock-in fluorescent reporters at the endogenous murine Snail1 and Snail2 (Slug) EMT-TF loci, we have shown that distinct EMT programs drive EMT during normal mammary gland development and mammary tumorigenesis [45]. In fact, Snail1-mediated EMT, but not Slug-mediated EMT, was activated at early stage of tumorigenesis and Zeb1 expression was associated with progression to high-grade carcinomas and metastasis [45]. Further, it is plausible that activation of an EMT program is involved in the re-awakening of previously dormant, disseminated tumours cells (DTCs), enabling these cells to generate macroscopic metastases [49]. As experimental models improve technically, the ability to study a movable target like the EMT program will be enhanced. Efforts to observe this type of cell plasticity in in vivo models in real time with single-cell resolution will surely prove to be highly informative [50].
6 Cell plasticity in metastasis and chemoresistance
A major unresolved issue in cancer research today is the development of chemoresistance, that is, how do tumours acquire resistance to therapies to which they were previously responsive? This type of tumour evolution is often observed when cancers recur or metastasize and is often complicated by the shortage of appropriate second-line treatment options for patients suffering from such clinical relapses. Consequently, an ability to prevent the development of acquired therapeutic resistance should preserve the sensitivity to initially employed, first-line treatment regimens, yielding in turn, increased patient progression-free survival if not cures.
In the context of understanding the development of chemoresistance, some have observed that the EMT, in addition to its aforementioned prometastatic properties, is also associated with the acquisition of elevated resistance to therapy, in which cells that have undergone a partial EMT, and even acquired tumour-initiating potential, are more resistant to conventional chemotherapy than their more epithelial counterparts [9, 10, 12–14]. Moreover, in clinical studies of neoadjuvant systemic endocrine and chemotherapy of breast cancer, patients with residual disease were found to have increased CD44(+)/CD24(−/low), mesenchymal markers and claudin-low signatures, suggesting that cell populations surviving after these therapies are enriched for subpopulations of cells with mesenchymal features and, quite possibly, tumour-initiating power [13]. To date, the mechanisms operating to promote therapeutic resistance remain largely elusive. In some cases EMT-TFs can promote homologous recombination (HR)-mediated DNA damage repair and the resulting resolution of DNA breaks [12, 51]. In other cases, cells that have undergone at least a partial-EMT program exhibit heightened resistance to apoptosis or an ability to pump out cytotoxic drugs [14, 52, 53]. Accordingly, the successful development of therapeutic agents that selectively target the mesenchymal derivatives of an EMT, including TICs [11], may prove to be an effective way to inhibit metastasis and cancer recurrence.
An alternative strategy to employing agents that selectively target and eradicate TICs that have undergone at least a partial EMT would be to force such cells to exit the TIC/quasimesenchymal state and return to a more differentiated, epithelial, non-TIC state. By doing so, the resulting epithelial non-TIC cells would presumably be more effectively targeted by conventional therapies. For example, preliminary findings suggest that activating protein kinase A (PKA) may represent one such way to force cells to undergo the MET and exit the EMT/TIC cell state [54].
Given the observed phenotypic plasticity of carcinoma cells, it also becomes plausible that use of chemo- or radiotherapeutic treatment protocols actively promotes the activation of previously latent EMT programs and a resulting shift of carcinoma cells to more mesenchymal states. Answers to this question may shed light on the issue of awakening of dormant metastases and associated clinical relapses. As an example of such phenotypic plasticity, recent evidence shows that circulating tumour cells (CTCs) in ER+ breast cancer are quite plastic, whereby HER2+ CTCs arise alongside HER2− CTCs in patients whose primary tumours are classified as HER2− [55]. Here, chemotherapy was shown to promote conversion of HER2+ CTCs to the HER2− chemotherapy-resistant state.
Along these lines, we have shown in populations of basal-type breast cancer cells, the epithelial non-TIC fraction shows remarkable plasticity, and in combination with the appropriate contextual signals in vivo, can undergo a partial EMT to generate de novo TICs [22, 56]. It will be interesting in the future to determine if the harsh conditions of the tissue microenvironment created by standard therapies promote metastatic outgrowth through programs such as the EMT. Interestingly, acting in the opposite direction, eribulin, a naturally derived mitotic spindle inhibitor that has been FDA-approved for use in advanced breast cancer and liposarcoma, has been shown to increase tumour perfusion through vascular remodelling in in vivo preclinical models of triple-negative breast cancer [57], and further, to suppress metastasis by reversing the EMT phenotype and promoting residence in the epithelial cell state [58].
7 Targeting the EMT as a therapeutic strategy in cancer
Few therapeutic clinical trials are currently underway that test the therapeutic efficacy of agents specifically designed to suppress expression of the EMT program. One promising agent is AB-16B5, a humanized monoclonal antibody directed against secreted clusterin (sCLU). sCLU has been shown to promote tumour cell survival and to be a potent stimulator of the EMT [59–61]. Thus, AB-16B5 is currently being evaluated in a phase I clinical trial in patients with solid malignancies. Monitoring of EMT and stem cell markers will be done as a secondary endpoint in peripheral blood circulating tumour cells and paired tumour biopsies (Clinicaltrials.gov identifier NCT02412462).
Notch inhibitors have been proposed to work through targeting stemness or the EMT, and there are currently a number of trials testing the efficacy of this class of drugs, with secondary endpoints evaluating changes in EMT markers in patients’ tumours. TGFβ inhibitors represent yet another class of therapies that have been investigated to target tumour cells that have activated versions of the EMT program. As discussed previously, TGFβ plays an important role in tumorigenesis and contributes to tumour cell proliferation, invasion and metastasis, inflammation, angiogenesis and escape from immune surveillance. The WNT/FZD signalling pathway is also implicated in tumour cell dedifferentiation and TIC function in numerous cancer types. Galunisertib, TEW-7197 and PF-03446962 are small molecule antagonists of the TGFβ receptor type 1 currently in early-phase clinical trials, either applied alone and in combination with other targeted therapies in various cancer types (Table 1). There are a number of Wnt inhibitors that have been developed that act via a variety of mechanisms. These include ETC-1922159, an inhibitor of the membrane-bound O-acyltransferase (MBOAT) porcupine (PORCN) in the endoplasmic reticulum, resulting in the inhibition of posttranslational palmitoylation of Wnt ligands and their secretion. OMP-54F28 is a recombinant fusion protein that binds WNT ligands and blocks WNT signalling, doing so through the extracellular domain of the Frizzled-8 receptor fused to a human IgG1 Fc fragment [62].
One of the challenges associated with evaluating biomarkers in clinical trials is the difficulty in obtaining pre- and posttreatment tissues to evaluate changes resulting from the administered therapy. Neoadjuvant trials, however, are particularly powerful, as they allow for the collection of paired samples from patients before and after treatment that are studied in order to evaluate directly the success of the agent under study; a number of therapies are currently being evaluated in this setting where evidence of the EMT is included as an endpoint (Table 1). One of these trials is eribulin (discussed above) followed by doxorubicin and cyclophosphamide in inflammatory breast cancer, which builds on preclinical findings and includes the secondary endpoints of evaluating the gene expression of 10-EMT-related genes in the posttreatment tumour samples in response to therapy (Clinicaltrials.gov identifier NCT02623972). Other clinical trials in the advanced setting that have included the evaluation of biomarkers of reduced EMT as a translational endpoint and include vorinostat, a histone deacetylase inhibitor, in combination with lapatinib in patients with advanced breast cancer Clinicaltrials.gov (identifier NCT01118975).
8 Summary
Arguably, the most important advance in the field of EMT and cancer metastasis is the knowledge that maintenance of cell plasticity elicits the most favourable outcome for successful completion of the metastatic cascade. Different lines of evidence support the idea that cancer cells can reside in various phenotypic states along the EMT spectrum, where cells can retain epithelial characteristics together with newly acquired mesenchymal ones. Importantly, the idea that cancer cells can transition dynamically between the epithelial, partial-EMT and mesenchymal states underlies their ability to adapt, survive and seed metastatic deposits. This type of phenotypic plasticity, governed by a combination of intrinsic and extrinsic factors, also endows cancer cells with tumour-initiating potential. In addition, the ability of carcinoma cells to maintain phenotypic plasticity may explain how such cells develop therapeutic resistance and drive cancer recurrence. In light of this knowledge, identifying the mechanisms that enable cell plasticity and the ability to accurately determine the prevalence of the EMT/MET in carcinoma cells should reveal new avenues for diagnosing disease severity and identifying the potential for progression and recurrence. Indeed, in this age of genomics coupled with the ability to analyse patient tissue with single-cell resolution, combining findings arising from clinical trials and leading-edge EMT research holds great promise for the future.
References
Hay, E. D. (2005). The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Developmental Dynamics, 233(3), 706–720.
Lamouille, S., Xu, J., & Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews. Molecular Cell Biology, 15(3), 178–196.
Peinado, H., Olmeda, D., & Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews. Cancer, 7(6), 415–428.
Thiery, J. P., Acloque, H., Huang, R. Y., & Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890.
Chaffer, C. L., & Weinberg, R. A. (2011). A perspective on cancer cell metastasis. Science, 331(6024), 1559–1564.
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., & Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America, 100(7), 3983–3988.
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133(4), 704–715.
Morel, A. P., Lievre, M., Thomas, C., Hinkal, G., Ansieau, S., & Puisieux, A. (2008). Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS One, 3(8), e2888.
Chen, J., Li, Y., Yu, T. S., McKay, R. M., Burns, D. K., Kernie, S. G., et al. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature, 488(7412), 522–526.
Kurrey, N. K., Jalgaonkar, S. P., Joglekar, A. V., Ghanate, A. D., Chaskar, P. D., Doiphode, R. Y., et al. (2009). Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells, 27(9), 2059–2068.
Gupta, P. B., Onder, T. T., Jiang, G., Tao, K., Kuperwasser, C., Weinberg, R. A., et al. (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell, 138(4), 645–659.
Zhang, P., Wei, Y., Wang, L., Debeb, B. G., Yuan, Y., Zhang, J., et al. (2014). ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature Cell Biology, 16(9), 864–875.
Creighton, C. J., Li, X., Landis, M., Dixon, J. M., Neumeister, V. M., Sjolund, A., et al. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America, 106(33), 13820–13825.
Smith, B. N., & Bhowmick, N. A. (2016). Role of EMT in metastasis and therapy resistance. J Clin Med, 5(2).
Nieto, M. A., Huang, R. Y., Jackson, R. A., & Thiery, J. P. (2016). EMT: 2016. Cell, 166(1), 21–45.
Bhowmick, N. A., Neilson, E. G., & Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature, 432(7015), 332–337.
Scheel, C., Eaton, E. N., Li, S. H., Chaffer, C. L., Reinhardt, F., Kah, K. J., et al. (2011). Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell, 145(6), 926–940.
Guo, W. (2014). Concise review: breast cancer stem cells: regulatory networks, stem cell niches, and disease relevance. Stem Cells Transl Med, 3(8), 942–948.
Wellner, U., Schubert, J., Burk, U. C., Schmalhofer, O., Zhu, F., Sonntag, A., et al. (2009). The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nature Cell Biology, 11(12), 1487–1495.
Kim, N. H., Kim, H. S., Li, X. Y., Lee, I., Choi, H. S., Kang, S. E., et al. (2011). A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. The Journal of Cell Biology, 195(3), 417–433.
Siemens, H., Jackstadt, R., Hunten, S., Kaller, M., Menssen, A., Gotz, U., et al. (2011). miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle, 10(24), 4256–4271.
Chaffer, C. L., Marjanovic, N. D., Lee, T., Bell, G., Kleer, C. G., Reinhardt, F., et al. (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell, 154(1), 61–74.
Siegel, P. M., & Massague, J. (2003). Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nature Reviews. Cancer, 3(11), 807–821.
Oft, M., Peli, J., Rudaz, C., Schwarz, H., Beug, H., & Reichmann, E. (1996). TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes & Development, 10(19), 2462–2477.
Muthusamy, B. P., Budi, E. H., Katsuno, Y., Lee, M. K., Smith, S. M., Mirza, A. M., et al. (2015). ShcA protects against epithelial-mesenchymal transition through compartmentalized inhibition of TGF-beta-induced Smad activation. PLoS Biology, 13(12), e1002325.
Gregory, P. A., Bracken, C. P., Smith, E., Bert, A. G., Wright, J. A., Roslan, S., et al. (2011). An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Molecular Biology of the Cell, 22(10), 1686–1698.
Nieswandt, B., Hafner, M., Echtenacher, B., & Männel, D. N. (1999). Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Research, 59(6), 1295.
Trikha, M., Zhou, Z., Timar, J., Raso, E., Kennel, M., Emmell, E., et al. (2002). Multiple roles for platelet GPIIb/IIIa and alphavbeta3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Research, 62(10), 2824–2833.
Labelle, M., Begum, S., & Hynes, R. O. (2011). Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell, 20(5), 576–590.
Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438(7069), 820–827.
Hiratsuka, S., Watanabe, A., Aburatani, H., & Maru, Y. (2006). Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nature Cell Biology, 8(12), 1369–1375.
Peinado, H., Lavotshkin, S., & Lyden, D. (2011). The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Seminars in Cancer Biology, 21(2), 139–146.
Castellana, D., Zobairi, F., Martinez, M. C., Panaro, M. A., Mitolo, V., Freyssinet, J. M., et al. (2009). Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Research, 69(3), 785–793.
Canesin, G., Cuevas, E. P., Santos, V., Lopez-Menendez, C., Moreno-Bueno, G., Huang, Y., et al. (2015). Lysyl oxidase-like 2 (LOXL2) and E47 EMT factor: novel partners in E-cadherin repression and early metastasis colonization. Oncogene, 34(8), 951–964.
Alix-Panabieres, C., Riethdorf, S., & Pantel, K. (2008). Circulating tumor cells and bone marrow micrometastasis. Clinical Cancer Research, 14(16), 5013–5021.
Gunasinghe, N. P., Wells, A., Thompson, E. W., & Hugo, H. J. (2012). Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Reviews, 31(3–4), 469–478.
Stankic, M., Pavlovic, S., Chin, Y., Brogi, E., Padua, D., Norton, L., et al. (2013). TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Reports, 5(5), 1228–1242.
Livasy, C. A., Karaca, G., Nanda, R., Tretiakova, M. S., Olopade, O. I., Moore, D. T., et al. (2006). Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Modern Pathology, 19(2), 264–271.
Rakha, E. A., Putti, T. C., Abd El-Rehim, D. M., Paish, C., Green, A. R., Powe, D. G., et al. (2006). Morphological and immunophenotypic analysis of breast carcinomas with basal and myoepithelial differentiation. The Journal of Pathology, 208(4), 495–506.
Bonnomet, A., Syne, L., Brysse, A., Feyereisen, E., Thompson, E. W., Noel, A., et al. (2012). A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene, 31(33), 3741–3753.
Schmidt, J. M., Panzilius, E., Bartsch, H. S., Irmler, M., Beckers, J., Kari, V., et al. (2015). Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Reports, 10(2), 131–139.
Tran, H. D., Luitel, K., Kim, M., Zhang, K., Longmore, G. D., & Tran, D. D. (2014). Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Research, 74(21), 6330–6340.
Tsai, J. H., Donaher, J. L., Murphy, D. A., Chau, S., & Yang, J. (2012). Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell, 22(6), 725–736.
Chaffer, C. L., Brennan, J. P., Slavin, J. L., Blick, T., Thompson, E. W., & Williams, E. D. (2006). Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Research, 66(23), 11271–11278.
Ye, X., Tam, W. L., Shibue, T., Kaygusuz, Y., Reinhardt, F., Ng Eaton, E., et al. (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature, 525(7568), 256–260.
Brabletz, S., & Brabletz, T. (2010). The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Reports, 11(9), 670–677.
Ye, X., & Weinberg, R. A. (2015). Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends in Cell Biology, 25(11), 675–686.
De Craene, B., & Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews. Cancer, 13(2), 97–110.
De Cock, J. M., Shibue, T., Dongre, A., Keckesova, Z., Reinhardt, F., & Weinberg, R. A. (2016). Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res.
Zhao, Z., Zhu, X., Cui, K., Mancuso, J., Federley, R., Fischer, K., et al. (2016). In vivo visualization and characterization of epithelial-mesenchymal transition in breast tumors. Cancer Research, 76(8), 2094–2104.
Cortez, M. A., Valdecanas, D., Zhang, X., Zhan, Y., Bhardwaj, V., Calin, G. A., et al. (2014). Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Molecular Therapy, 22(8), 1494–1503.
Moitra, K. (2015). Overcoming multidrug resistance in cancer stem cells. BioMed Research International, 2015.
Abdullah, L. N., & Chow, E. K. H. (2013). Mechanisms of chemoresistance in cancer stem cells. Clinical and Translational Medicine, 2, 3.
Pattabiraman, D. R., Bierie, B., Kober, K. I., Thiru, P., Krall, J. A., Zill, C., et al. (2016). Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science, 351(6277) aad3680.
Jordan, N. V., Bardia, A., Wittner, B. S., Benes, C., Ligorio, M., Zheng, Y., et al. (2016). HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature, 537(7618), 102–106.
Chaffer, C. L., Brueckmann, I., Scheel, C., Kaestli, A. J., Wiggins, P. A., Rodrigues, L. O., et al. (2011). Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proceedings of the National Academy of Sciences of the United States of America, 108(19), 7950–7955.
Funahashi, Y., Okamoto, K., Adachi, Y., Semba, T., Uesugi, M., Ozawa, Y., et al. (2014). Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Science, 105(10), 1334–1342.
Yoshida, T., Ozawa, Y., Kimura, T., Sato, Y., Kuznetsov, G., Xu, S., et al. (2014). Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. British Journal of Cancer, 110(6), 1497–1505.
Wang, C., Jiang, K., Kang, X., Gao, D., Sun, C., Li, Y., et al. (2012). Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. The International Journal of Biochemistry & Cell Biology, 44(12), 2308–2320.
Tremblay, G., Malouin, M., Grothe, S., Kalbakji, A., Roy, S., Pagé, M., et al. (2010). Abstract 1467: AB-16B5, a therapeutic monoclonal antibody against human clusterin that blocks the epithelial-to-mesenchymal transition. Cancer Research, 70(8 Supplement), 1467 [10.1158/1538-7445.AM10-1467].
Tremblay, G. B., Viau, E., & Filion, M. (2012). Abstract LB-297: the EMT inhibitor AB-16B5 interacts with specific isoforms of secreted clusterin. Cancer Research, 72(8 Supplement) LB-297. [10.1158/1538-7445.AM2012-LB-297].
Jimeno, A., Gordon, M. S., Chugh, R., Messersmith, W. A., Mendelson, D. S., Dupont, J., et al. (2014). Abstract 2505; a first-in-human phase 1 study of anticancer stem cell agent OMP-54F28 (FZD8-Fc), decoy receptor for WNT ligands, in patients with advanced solid tumors. Journal of Clinical Oncology, 32, 5s.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chaffer, C.L., San Juan, B.P., Lim, E. et al. EMT, cell plasticity and metastasis. Cancer Metastasis Rev 35, 645–654 (2016). https://doi.org/10.1007/s10555-016-9648-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-016-9648-7