
Abstract
The mammalian target of rapamycin (mTOR) was reported to regulate cell autophagy and outcomes of several neurological diseases. Mitochondria, which serve as critical organelles in neurons. are also involved in the pathology of neurological diseases. However, the role of mTOR in mitochondrial morphology has not been clarified especially in subarachnoid hemorrhage (SAH). In this study, we established SAH models both in vivo and in vitro. Rapamycin and 3-methyl adenine (3-MA) were then administered to alter mTOR activity. Post-SAH assessment included SAH grading, neurological evaluation, blood–brain barrier (BBB) permeability, brain water content, mitochondrial membrane potential (MMP), mitochondrial morphology, ATP content, cell viability, cytotoxicity, and expression of proteins related to apoptosis and mitochondrial fission. The results showed that (1) neurological deficits, BBB permeability, and brain edema were increased after SAH and that cell viability was exacerbated in brain tissue. (2) Excessive mitochondrial fission was evident based on changes in mitochondrial morphology, while MMP and ATP content were decreased in neurons after SAH. (3) Administration of rapamycin improved the excessive mitochondrial fission and restored mitochondrial function, which subsequently reduced apoptosis. (4) 3-MA showed an adverse effect on mitochondria and aggravated excessive mitochondrial fission and dysfunction in SAH. Neurological deficits and neuronal viability were also exacerbated following the administration of 3-MA. Therefore, our study suggests that mTOR inhibition has neuroprotective effects against neuronal injury after SAH via alleviating excessive mitochondrial fission.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Subarachnoid hemorrhage (SAH) is a subtype of hemorrhagic stroke with a high mortality and morbidity (Sun et al. 2014; Zhao et al. 2019). Despite therapies and improved clinical management, patients still suffer lasting psychological and physical effects even after “good” outcomes (Schuette and Barrow 2013). Early brain injury (EBI) and cerebral vasospasm are two major complications after SAH. Outcomes of anti-vasospasm treatment are poor, and researchers consider EBI that has occurred within 72 h after SAH as the primary cause of these poor outcomes after SAH (Sun et al. 2019; Zhao et al. 2019). The pathologic mechanisms of EBI include increased intracranial pressure, decreased perfusion pressure, impaired blood–brain barrier (BBB), brain ischemia, brain edema, and toxicity from hemolysate. All of these pathological processes ultimately result in neuronal injury and death. In addition, mitochondrial dysfunction is involved in nearly all of these pathologic mechanisms (Cahill et al. 2006; Wang et al. 2012).
Mitochondria are abundant in neurons due to their complex functions and high demand for energy. Therefore, mitochondria play a pivotal role in both physiological and pathological events during neurological disorders (Cheng et al. 2010). Many pathological mechanisms post-SAH, such as brain edema, oxidative stress, and calcium overload, are accompanied by mitochondrial dysfunction and often result in neuronal apoptosis. Alterations in morphology have become a hot topic in the study of mitochondrial dysfunction during cell apoptosis (Ikeda et al. 2014). Mitochondria are dynamic organelles that continuously strike a structural balance of fission and fusion. Dynamin-related protein 1 (Drp1) and fission protein 1 (Fis1) are reported to regulate mitochondrial fission, whereas mitochondrial fusion is regulated by optic atrophy 1 and the mitofusins (Mfn). Usually, DRP-1 is activated by phosphorylation (p-Drp1), whereby p-Drp1 assembles at the mitochondrial surface similar to annuluses and constricts both inner and outer membranes with energy from hydrolyzing guanosine-5′-triphosphate (GTP), after which the mitochondrion undergoes fission to generate multiple mitochondria (Zhang et al. 2013). Abnormalities in mitochondrial morphology, especially excessive fission, has been reported in many nervous system diseases (Knott and Bossy-Wetzel 2008). In an ischemic study, researchers found that Drp1 and related downstream proteins were increased immediately after ischemia, especially in the penumbral region (Pradeep et al. 2014). Our previous work also showed that mitochondria in the brain of rats appeared to undergo excessive fission after SAH, and inhibition of mitochondrial fission facilitated the improvement of neurological deficits and neuronal apoptosis (Wu et al. 2017, Zhang et al. 2018). Emerging evidence indicates that an imbalance of mitochondrial morphology is involved in the pathology of neurological disorders and might serve as a potential therapeutic target.
The mammalian target of rapamycin (mTOR) is known as a key regulator of autophagy (Lee et al. 2009). Autophagy is a catabolic process of degrading intracellular components such as damaged proteins and organelles (Kardideh et al. 2019). Several studies found that autophagy was markedly enhanced at the early stage of EBI and had neuroprotective effects against apoptosis in many neurodegenerative diseases (Jing et al. 2012, Guo et al. 2018). mTOR activity was shown to negatively regulate autophagy; the mTOR inhibitor, rapamycin, upregulated autophagy, whereas 3-methyl adenine (3-MA), an agonist of mTOR, inhibited autophagy (Hu et al. 2018, Kardideh et al. 2019). The significance of the mTOR pathway has been underscored in a myriad of nervous system disorders (Ma et al. 2015, Shi et al. 2017). In a study of SAH, researchers found that rapamycin could inhibit mTOR activity via suppressing its phosphorylation, which then attenuated neuronal injury in EBI (You et al. 2016). Another study on acute focal brain damage found that rapamycin, in addition to stimulating autophagy, could promote Drp1 accumulation and mitochondrial fission, which facilitated the degradation of damaged mitochondria by autophagy (Cavallucci et al. 2014). In rats subjected to hypoxia–ischemia, rapamycin was reported to prevent necrotic cell death via the mitochondrial pathway, while administration of 3-MA after rapamycin elicited the translocation of Bad and Bax to the mitochondria, which increased the cell death (Carloni et al. 2012). In a study of Leigh syndrome, Johnson et al. found that inhibition of mTOR-alleviated mitochondrial dysfunction in mouse models (Johnson et al. 2013). Unfortunately, there are no correlative reports regarding the relationship between mTOR, mitochondrial function, and mitochondrial morphology after SAH.
In this study, we used SAH models, both in vivo and in vitro, to detect abnormalities-associated mitochondrial function and morphology after SAH. After, we administrated rapamycin and 3-MA to explore the effect of mTOR activity on mitochondrial abnormalities after SAH. The present study might provide evidence on an effective downstream pathway of mTOR to reduce neuronal apoptosis after SAH.
Materials and Methods
Animals and SAH Models In Vivo
Male Wistar rats weighing 250–350 g were purchased from the Animal Center of the Second Affiliated Hospital of Harbin Medical University (Harbin. China). The animals were acclimated at the animal center for at least 3 days before the operation. All experimental procedures were approved by the Animal Care and Use Committee of the First Affiliated Hospital of Harbin Medical University. The protocols for the animal experiments were performed in accordance with the Care and Use of Laboratory Animals published by the U.S. National Institution of Health.
The endovascular perforation method was used on rats to establish SAH models in this study. As in our previous report (Wu et al. 2017, Zhang et al. 2018), animals were anesthetized with pentobarbital sodium (50 mg/kg) by intraperitoneal injection. The external carotid artery (ECA), internal carotid artery (ICA) and common carotid artery were fully exposed, and after distal ligation of ECA, a blunt 4-0 nylon suture was placed into the ECA and advanced through the ICA for about 2.0 cm. Prior to extraction, the nylon suture was held motionless for approximately 10 s to ensure the successful puncture of the artery. For the sham-operated animals, all procedures were same except for the puncture of the ECA. At 24 h post-SAH, surviving animals were anesthetized and perfused with ice-cold saline, then brain tissue samples were removed and stockpiled in liquid nitrogen for subsequent testing.
Oxyhemoglobin (Hb)-Induced Neuronal Injury
The mouse hippocampal neuronal cell line, HT22, purchased from Biotechnology Co. Ltd. Shanghai enzyme research (Shanghai, China), was used in the present study. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (HyClone, Logan, UT, USA) containing 10% fetal bovine serum (HyClone) at 37 °C in a 5% CO2 humidified atmosphere.
As in previous reports (Sun et al. 2014, Zhang et al. 2018), Hb (Sigma-Aldrich, MO, USA) co-incubation was used to induce SAH in vitro. In the Hb group, Hb was dissolved in complete culture medium and filtered through a 0.22-μm sterile filter. HT22 cells were then co-incubated with Hb (10 μM) for 24 h.
Experimental Groups
As shown in Fig. 1, a total of 147 animals were randomly divided into five groups: the sham group, where animals underwent sham-operation; and the SAH, SAH + vehicle, SAH + rapamycin, and SAH + 3-MA groups, where animals were all subjected to SAH. Animals in the SAH + vehicle group were administrated with 2% dimethylsulfoxide (DMSO) in normal saline at 30 min post-SAH. In the SAH + rapamycin and SAH + 3-MA groups, animals were respectively administrated with 40 µl rapamycin (1 ng/µl, in 2% DMSO) and 50 µl 3-MA (10 mM, Sigma) intraventricularly at 30 min post-SAH. Animals that died during surgery and within 24 h after SAH were not counted. All surviving animals were sacrificed at 24 h after SAH. The dose and concentration of rapamycin and 3-MA were selected based on a previous study (Zhao et al. 2013).

Experimental groups and protocols. MTT 3-(4,5-dimethyl-2-thiazolyl, MDA malondialdehyde, TEM transmission electron microscopy, WB western blot
For the in vitro experimental design, the cells were also divided into five groups: the control group, where cells were only incubated with complete culture medium; the Hb group, where cells were co-incubated with 10 μM Hb in complete culture medium for 24 h; the Hb + vehicle group, where cells were pretreated with 2% DMSO in 10 μM Hb in complete culture medium for 24 h; and the Hb + rapamycin and Hb + 3-MA groups, where cells were respectively co-incubated with 100 nM rapamycin and 5 mM 3-MA (diluted with 1% DMSO in 10 μM Hb in complete medium; Sigma-Aldrich) for 24 h, the dose and time point of Hb were also selected based on previous studies (Li et al. 2017, Zhang et al. 2018).
SAH Grading
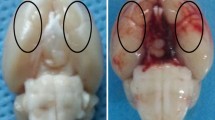
The severity of SAH was estimated by two blinded researchers following guidelines from a previous report (Sugawara et al. 2008). As shown in Fig. 2a, after the rats were sacrificed at 24 h post-SAH and perfused with ice-cold saline, the basal brain was divided into six segments. Each segment was scored with a grade from 0 to 3 according to the amount of blood clotting. The total score of the six segments ranged from 0 to 18, and revealed the bleeding scale of every SAH model. The higher scores indicate more blood clotting.

Representative images of sham-operated and SAH-operated brains, SAH grade scores, and mortality of rats. a The basal surface of the brain was divided into six areas for the SAH grade scoring. b Representative image of a sham-operated brain. c Representative image of a SAH-operated brain and schematic representation of the area for the cortex samples. d No significant difference in the SAH grade was found among the SAH rats in the different groups (n = 24, p > 0.05). e The mortality of each group
Neurological Evaluation and Brain Water Content
As in previous reports, the Garcia JH scores were used to evaluate neurological deficits, the neurological scores were evaluated by two blinded observers at 24 h post-SAH (Wang et al. 2012, Wu et al. 2017). Briefly, the neurological deficits consisted of six tests, including spontaneous activity, symmetry of the four limbs, forepaw outstretching, climbing, body proprioception and response to vibrissae touch. According to detailed standards, each test was classified from 1 to 3 or 0 to 3. The lowest score was 6 and the highest was 18. The higher scores indicate better neurological outcomes.
The dry–wet weight method was used to test brain water content after SAH. The brains were removed at 24 h after the operation and divided into four parts: the left hemisphere, right hemisphere, brain stem, and cerebellum. Each part was weighed (wet weight), then dried in an oven for 24 h at 105 °C (dry weight). The percentage of brain water was calculated as (wet weight − dry weight)/wet weight × 100%.
BBB Permeability
BBB permeability was assessed as previously reported (Teng et al. 2016). Briefly, at 24 h post-SAH, Evan’s blue dye (2%, 5 mL/kg, Sigma-Aldrich) was slowly injected into the right femoral vein. After the dye was circulated for 60 min, the rats were perfused with phosphate-buffered saline (PBS). The brain samples were removed and homogenized in 50% trichloroacetic acid. The amount of transudatory Evan’s blue dye was determined using spectroflourophotometry. Measurements were conducted at an excitation wavelength of 620 nm. All results were normalized to the sham group.
TUNEL Stain and MTT Assay
The cortex from the basal surface of the rat brain was selected to detect neuronal apoptosis by TUNEL stain. Paraformaldehyde-fixed (4%) brain tissues were embedded in paraffin and sectioned (4 μm thick) with a microtome. According to protocols of the manufacturer, the sections were stained with TUNEL-staining Kit (Roche, Basel, Switzerland) and examined by an LSM-710 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY, USA). After positive cells were identified and counted by a blinded investigator, the apoptosis index was calculated with the average number of TUNEL-positive cells in six microscopic fields in three different slices of the brain.
The MTT assay (Sigma-Aldrich) was used to detect the cell viability in each group. Briefly, 2 × 103 cells in 100 μL complete medium were seeded into a 96-well plate. After the co-incubation, MTT solution (5 mg/mL, 20 μL) was added to the cells and incubated for 4 h at 37 °C. DMSO (150 μL) was added to each test well and incubated for 20 min at 37 °C. The optical density (OD) was read on a microplate reader (BioTek, VT, USA) at 490 nm. Each test was done in triplicate wells and the result was repeated six times for each test group.
Transmission Electron Microscopy (TEM)
TEM was used to observe the change of mitochondrial structure in each group. Brains of rats were minced into small fragments (< 1 mm3), and fixed with 2.5% buffered glutaraldehyde for 4 h at 4 °C. Fragments were then fixed with 2% osmic acid in the fixative above for 90 min, dehydrated with incremental concentrations of ethyl alcohol and embedded in araldite for 24 h at 60 °C. The polymerized specimens were cut into 60–70 nm slices with an EM UC7 ultramicrotome (Leica, Wetzlar, Germany). After fixation to nickel grids, the sections were stained with uranyl acetate and lead citrate. Microscope photographs were taken with a transmission electron microscope (Carl Zeiss).
Malondialdehyde (MDA) Measurement
MDA levels in the brain were measured with Lipid Peroxidation MDA Assay Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The left hemisphere of brains in different groups was used for the measurement of MDA. A spectraMax M2 spectrometer (Molecular Devices) was used to calculate data. The level of MDA was expressed as nmol/mg protein (λ = 523 nm),
ATP Content and Mitochondria Extraction
ATP content in HT22 cells was luminometrically determined by a rapid bioluminescent ATP assay kit (EnzyLight, USA). The ATP content was assessed according to the manufacturer’s protocol. The luminescence in each group was measured on a PowerWave 340 microplate spectrophotometer (Bio-Tek, Winooski, USA). The results were normalized to the control group, the results were repeated six times per group.
Mitochondria extracted from brain tissues were used to explore the expression of apoptotic proteins. The brains were immediately removed after perfusion with ice-cold saline. The cerebral cortex covered with the blood clot was then isolated and frozen in liquid nitrogen. Mitochondrial fractions were isolated with the Mitochondrial Extraction Kit (Solarbio Life Sciences, Beijing, China). According to the manufacturer’s instructions, the mitochondrial fraction was extracted and resuspended in store buffer to store at − 80 °C, the supernatants obtained were stored as cytosol fractions.
Super-Resolution Imaging Microscope
To further evaluate the change of mitochondrial morphology, HT22 cells were seeded on a glass-bottom dish (Shengyou Biotechnology, Hangzhou, China) and incubated for 24 h. After the co-incubation, the cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. Afterward, cells were washed with PBS twice and were labeled with Mito Tracker Red CM-XRos (Invitrogen, Camarillo, CA, USA) for 30 min at 37 °C. Then the mitochondria in cells were observed using a DeltaVision OMX super-resolution imaging microscope (GE, Atlanta, GA, USA). Mitochondrial morphology in cells was evaluated as previously reported (Regmi et al. 2014). The mitochondrial length, circularity, and area were analyzed by measuring the fluorescent area with the Image J software (National Institutes of Health, Bethesda, MD, USA). Mitochondrial circularity is a measurement of “roundness” of mitochondria with 0 referring to a straight line and 1 referring to a perfect circle. The results were repeated six times per group.
Flow Cytometry
Flow cytometry was used to detect the cell apoptosis rate and mitochondrial MMP in each group. The apoptosis index was tested with the AnnexinV:FITC Apoptosis Detection Kit (BD Pharmingen, Franklin Lakes, NJ, USA). According to the protocol, the cells were stained with Annexin V-FITC dye and PI dye without light for 30 min at 37 °C, after which the cells were washed with warm PBS once. The resuspended cells in PBS were then subjected to flow cytometry (Beckman Coulter, USA). CellQuest software (BD Biosciences) was used to quantify the number of apoptotic cells.
The MitoProbe JC-1 Assay Kit (Life Technologies, Carlsbad, CA, USA) was used to detect the MMP in cells. Briefly, the cells in each group were collected and resuspended with warm PBS at approximately 1 × 106 cells/mL. For the control group, 1 μL CCCP (50 mM) was added and incubated for 5 min at 37 °C. Cells in the other groups were incubated with 10 μL JC-1 (200 μM) for 30 min at 37 °C. Prior to being analyzed on the cytometer, cells were washed with warm PBS and resuspended with 500 μL PBS (Mendelev et al. 2011). The CCCP-treated sample was used as standard compensation.
All experiments were replicated at least six times. The results were analyzed by FlowJo software (Tree Star, Ashland, OH, USA).
Western Blot
Western blot was used to determine the protein expression of apoptosis and mitochondrial fission in mitochondria and the cerebral cortex. Briefly, the samples (50 μg) were loaded onto PAGE gels (Genscript, Piscataway Township, NJ, USA), and following electrophoresis, were transferred to nitrocellulose membranes (Life Technologies) and blocked with 5% nonfat milk for 1 h at 37 °C. The membranes were then incubated with the following primary antibodies: anti- Cytochrome C (Cyt C), anti-cleaved caspase-3 and anti-β-actin (1:1000, Beyotime Biotechnology, Shanghai, China); and anti-COX IV, p-DRP1 (1:1000, Cell Signaling Technology, Boston, MA, USA) and DRP1 (1:1000, Abcam, Cambridge, UK). The next day, the membranes were incubated with anti-rabbit or anti-mouse HRP-conjugated second antibodies for 1 h at room temperature and detected with the Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE, USA), The results were analyzed with the Image J software. β-Actin and COX IV were used as internal standards.
Statistical Analysis
All data were expressed as mean ± SD. Multiple comparisons were performed with one-way ANOVA; Bonferroni or Dunn’s post hoc analysis was used to determine where differences occurred. Differences in mortality between groups were tested with Fisher’s exact test. GraphPad Prism 6 (GraphPad, La Jolla, CA) was used in statistical analysis. p < 0.05 is considered statistically significant,
Results
Mortality and SAH Grade
Representative images of the brains of SAH-operated and sham-operated animals are shown in Fig. 2b and C. Blood clots are clearly evident on the basal surface and around the arteries of the brain in the SAH group. The SAH grading method (Fig. 2a), depending on the amount of blood clotting, was used to evaluate the severity of SAH. The mean of SAH grading score was 0 in the sham group. There was no difference among any of the experimental groups (SAH + vehicle, SAH + rapamycin, and SAH + 3-MA groups; p > 0.05, Fig. 2d).
A total of 123 animals underwent the SAH operation and 24 animals underwent the sham operation; a total of 96 animals survived after the SAH procedure. The mortality was 0% (0/24) in the sham group, 25.0% (8 of 32) in the SAH group, 27.3% (9 of 33) in the SAH + vehicle group, 14.3% (4 of 28) in the SAH + rapamycin group, and 33.4% (12 of 36) in the SAH + 3-MA group. In addition, most deaths occurred within 6 h after the operation. There was no significant difference between the SAH group and the SAH + vehicle group in regard to mortality (p > 0.05). Despite the fact that rapamycin and 3-MA, respectively, decreased and increased the mortality after SAH, there was also no statistical significance as compared with the SAH group (p > 0.05, Fig. 2e).
mTOR Inhibition Improved Neurological Deficits, BBB Permeability, and Brain Edema After SAH
Neurological deficits were evaluated with the Garcia JH score. Neurological function deteriorated in rats of the SAH group (vs. sham, p < 0.01), while administration of rapamycin and 3-MA, respectively, improved (vs. SAH, p < 0.01) and further aggravated (vs. SAH, p < 0.05) neurological deficits after SAH (Fig. 3a). There was no statistical significance between the SAH and the SAH + vehicle group. This result indicated that inhibition of mTOR-improved neurological outcomes of rats in the SAH group.

Inhibition of mTOR-alleviated neurological deficits and decreased BBB permeability and brain edema after SAH. a Neurological scores of surviving animals were evaluated at 24 h after the operation (n = 24). b Quantification of Evan’s blue extravasation was used to detect BBB permeability (n = 3). c The brain water content of different parts was used to detect the brain edema in each group (n = 6, **p < 0.01 vs. sham, #p < 0.05 and ##p < 0.01 vs. SAH)
Evan’s blue leakage was used to detect the BBB permeability. The result showed that the leakage was increased in the SAH group (vs. sham, p < 0.01) and vehicle group (vs. sham, p < 0.01). However, the leakage was decreased in the SAH + rapamycin group (vs. SAH, p < 0.05) and further increased in the SAH + 3-MA group (vs. SAH, p < 0.01, Fig. 3b). This result indicated mTOR inhibition improved BBB permeability after SAH.
Brain water content is known to reveal the severity of brain edema. The results showed that brain edema was obviously increased in the SAH group in the left (80.88 ± 1.38%, vs. sham, p < 0 0.01) and right hemispheres (80.62 ± 1.22%, vs. sham, p < 0.01), while there was no brain edema found in the cerebellum or brainstem. Brain edema in the rats of the vehicle group in both the left (80.78 ± 1.08%) and right hemispheres (80.65 ± 1.21%) was comparable to the SAH group (p > 0.05). Compared with the SAH group, administration of rapamycin reduced the brain water content in both the left (79.55 ± 0.99%, p < 0.05) and right hemispheres (79.13 ± 0.85%, p < 0.05), while administration of 3-MA obviously reduced the brain water content only in the left hemisphere (82.20 ± 0.50%, p < 0.05, Fig. 3c). Inhibition of mTOR-ameliorated brain edema after SAH.
mTOR Inhibition Ameliorated Neuronal Apoptosis After SAH
The TUNEL-staining method was used to detect apoptotic neurons following surgery. There were few TUNEL-positive cells in the sham-operated rat brains (6.5 ± 4.2%). Apoptotic cells were significantly increased in the SAH (39.6 ± 8.3%, vs. sham, p < 0.01) and SAH + vehicle groups (33.8 ± 8.3%, vs. sham, p < 0.01). Administration of rapamycin remarkably decreased apoptosis (16.5 ± 8.0%, vs. SAH, p < 0.01), but in the SAH + 3-MA group, apoptosis was significantly increased (55.4 ± 13.3%, vs. SAH, p < 0.05, Fig. 4a, b). Expression of cytoplasmic cleaved caspase-3 and Cyt C, both markers of apoptosis, were measured by western blot. Both proteins were upregulated in the SAH group (vs. sham, p < 0.05) and SAH + vehicle group (vs. sham, p < 0.05). Rapamycin significantly reduced the overexpression of cleaved caspase-3 (vs. SAH, p < 0.05) and Cyt C (vs. SAH, p < 0 0.05), while mTOR activation via 3-MA further upregulated the levels of both proteins (vs. SAH, p < 0.01, Fig. 4c–e).

Inhibition of mTOR-alleviated neuronal apoptosis after SAH. a Representative photographs of TUNEL staining in brain sections (scale bar = 25 μm). b Apoptosis index of the TUNEL staining in each group (n = 3). c Representative images of Cyt C and cleaved caspase-3 bands in rat neurons. d Quantification of the cleaved caspase-3 expression (n = 6). e Quantification of the Cyt C expression (n = 6). The results were normalized to β-actin. f The cell viability in each group was detected by the MTT assay (n = 6). g The apoptotic rate was assessed by flow cytometry. h Statistical analysis of the apoptotic rate in each group (n = 6, **p < 0.01 vs. sham/control, #p < 0.05 and ##p < 0.01 vs. SAH/Hb)
Cell viability and apoptosis in the Hb-treated cell models were also determined. MTT results showed that the cell viability in the Hb and Hb + vehicle groups was decreased when compared with the control group (p < 0.01), while the viability was increased after rapamycin treatment (vs. Hb, p < 0.05) and was decreased after 3-MA administration (vs. Hb, p < 0.05, Fig. 4f). Apoptosis in the SAH models in vitro was evaluated by the Annexin V/PI double stain, and the apoptotic cell populations were quantified by flow cytometry. The results showed that the mean proportion of apoptotic cells was increased from 13.19 ± 0.99% in the control group to 34.53 ± 1.04% in the Hb group (vs. control, p < 0.01) and 33.86 ± 2.57% in the Hb + vehicle group (vs. control, p < 0.01). Rapamycin treatment decreased the apoptosis rate to 18.43 ± 1.76% (vs. Hb, p < 0.01), while 3-MA treatment increased the apoptosis rate to 43.34 ± 1.57% (vs. Hb, p < 0.05, Fig. 4g, h). The data revealed that inhibiting mTOR with rapamycin facilitated the survival of HT22 cells treated with Hb.
mTOR Inhibition Improved Mitochondrial Dysfunction After SAH
The mitochondrial ultrastructure was observed using TEM. The micrographs showed that mitochondria in neurons of SAH rats were swollen, and the organelle structure including the crista was destroyed. Inhibiting mTOR with rapamycin ameliorated mitochondrial injury and improved ultrastructure. However, mitochondria in the SAH + 3-MA group were extremely swollen, and almost no ultrastructure was observed inside the mitochondria (Fig. 5a).

Inhibition of mTOR alleviated the mitochondrial dysfunction after SAH. a Representative micrographs of mitochondria observed by TEM. b Representative images of Cyt C band in mitochondria. c Quantification of the expression of Cyt C in mitochondria. (n = 6). d MDA levels in the brains were used to detect oxidative damage in each group (n = 6). e ATP content in cells of each group was used to reveal mitochondrial function (n = 6). f MMP in the cells was indicative of mitochondrial function in every group (n = 6, **p < 0.01 vs. sham/control, #p < 0.05 and ##p < 0.01 vs. SAH/Hb)
The expression of Cyt C in mitochondria was measured by western blot (Fig. 5b). Quantitative analysis showed Cyt C was downregulated in the mitochondria of the SAH group (vs. sham, p < 0.01) and SAH + vehicle group (vs. sham, p < 0 0.01). Administration of rapamycin partially upregulated mitochondrial Cyt C when compared with the SAH group (p < 0.05), while 3-MA increased downregulation of Cyt C (p < 0.05, Fig. 5c). The data indicated rapamycin inhibited the release of mitochondrial Cyt C into the cytoplasm. As an oxidative damage marker, MDA was measured in the brain of rats. MDA levels in the brains of rats in the SAH and SAH + vehicle groups were increased compared with those in the sham group (p < 0.01). Administration of rapamycin and 3-MA, respectively, reduced and increased MDA levels (vs. SAH, p < 0.05 for both, Fig. 5d).
ATP content and MMP in HT22 cells were used to detect mitochondrial function. The data of co-incubation groups were all normalized to the control group. After co-incubation with Hb, the ATP content in HT22 cells decreased (vs. control, p < 0.01). Compared with the Hb group, administration of rapamycin elevated the ATP content (p < 0.05); however, 3-MA further decreased the ATP content (p < 0.05, Fig. 5e). MMP in HT22 cells was detected with the JC-1 fluorescent probe. The results showed that MMP in the cells of the Hb and Hb + vehicle groups obviously depolarized (Fig. 5f), which indicated MMP in the Hb and vehicle groups significantly decreased (p < 0.01, vs. control), whereas, MMP in the Hb + rapamycin group and Hb + 3-MA groups, was respectively improved (p < 0.01) and deteriorated (p < 0.01) when compared with the Hb group. These results verified the inhibition of mTOR-ameliorated mitochondrial dysfunction after SAH.
mTOR Inhibition-Alleviated Excessive Mitochondrial Fission After SAH
Super-resolution imaging and expression of Drp1, as well as p-Drp1 were used to evaluate the change of mitochondrial morphology. Mitochondria were characterized as punctiform shape or linellae under the super-resolution imaging (Fig. 6a). The mitochondrial length, circularity, and area were measured in our study (Fig. 6b–d). Long and circular mitochondria could be observed in the micrographs of the control group. However, reduced mitochondrial length and circularity were found in the Hb (vs. control, p < 0.01) and Hb + vehicle groups (vs. control, p < 0.01). Mitochondrial area was also increased in the Hb (vs. control, p < 0.05) and Hb + vehicle groups (vs. control, p < 0.01). In the Hb + rapamycin group, linear and circular mitochondria appeared to be increased (vs. Hb, p < 0.05) and the mitochondrial area was decreased (vs. Hb, p < 0.05), whereas 3-MA further decreased the mitochondrial length (vs. Hb, p < 0.05), circularity (vs. Hb, p < 0.05) and increased the area (vs. Hb, p < 0.01) in the Hb-treated cells.

Inhibition of mTOR-alleviated excessive mitochondrial fission after SAH. a Representative micrographs observed by super-resolution microscopy showed the mitochondrial morphology in each group (scale bar = 10 μm). b Quantification of mitochondrial length in each group (n = 6). c Quantification of mitochondrial circularity in each group (n = 6). d Quantification of mitochondrial area in each group (n = 6). e Representative bands of Drp1 and p-Drp1. f Quantification of the expression of Drp1 in rat brains (n = 6). g Quantification of the expression of p-Drp1 in rat brains (n = 6). Results were normalized to β-actin (**p < 0.01 vs. sham/control, #p < 0.05 and ##p < 0.01 vs. SAH/Hb)
The expression of Drp1 and p-Drp1 was used to quantify mitochondrial fission. Both Drp1 and p-Drp1 expression in the cortex of rats was increased after SAH (vs. sham, p < 0.01). Compared with the SAH group, Drp1 and p-Drp1 were downregulated after rapamycin administration (p < 0.01), while the two proteins were upregulated in the SAH + 3-MA group (vs. SAH, p < 0.05, Fig. 6e–g). The results suggest that mitochondria in neurons undergo excessive fission after SAH, and mTOR inhibition-reduced morphological damage.
Discussion
In the present study, mitochondrial morphology and function in neurons were found to be deteriorated in our SAH models, which may have contributed to EBI after SAH. We next tested the effect of mTOR activity on these mitochondrial abnormalities. The results indicated that inhibition of mTOR with rapamycin could alleviate excessive mitochondrial fission and dysfunction in EBI and improve neurological deficits after SAH. In contrast, the mTOR agonist, 3-MA, further aggravated mitochondrial abnormalities. This work provides a new direction in the therapy of SAH involving the restoration of mitochondrial morphological homeostasis.
Mitochondria are dynamic organelles that constantly undergo fission and fusion under physiological conditions. Researchers verified that the fusion and fission homeostasis acted as a key checkpoint for mitochondrial functionality (Mendl et al. 2011). In the process of disease, aging, and development, the morphological balance was deemed to be crucial for mitochondrial function (Chan 2006). Mitochondrial fission in the brain of rats was distinctly increased after ischemic injury, while inhibition of excessive fission was shown to reduce mitochondrial dysfunction and apoptosis in neurons (Zhang et al. 2013; Tian et al. 2014). Studies on EBI after SAH verified mitochondrial dysfunction in neurons, which included cellular calcium overload, oxidative stress injury, and release of pro-apoptosis factors (Cahill et al. 2006; Xu et al. 2015). Our previous work indicated that excessive mitochondrial fission might be the cause of mitochondrial dysfunction, and that a fission inhibitor relieved mitochondrial injury after SAH (Wu et al. 2017). As a main regulatory factor of fission, the active form of Drp1, p-Drp1, assembles on the outer membrane of mitochondria and “cleaves” mitochondria with energy utilized from GTP hydrolyzation (Liu et al. 2012; Zhang et al. 2013). Therefore, the expression of Drp1, and in particular p-DRP1, revealed the extent of mitochondrial fission. Researchers have identified Drp1 and p-Drp1 as biomarkers of mitochondrial fission after stroke (Liu et al. 2012). In our study, results from TEM indicated the mitochondrial impairment, and the expression of both Drp1 and p-Drp1 were upregulated after SAH, These were in agreement with our previous report (Wu et al. 2017). We then further investigated mitochondrial dysfunction and morphological abnormalities in HT22 cells treated with Hb. MMP depolarization is known to play a key role in the release of pro-apoptotic proteins including Cyt C from mitochondria into the cytoplasm (Mendl et al. 2011). In addition, mitochondria are the major sources of generating ATP in neurons (Chan 2006). The population of cells with depolarized MMP significantly increased while the ATP content decreased in cells treated with Hb. Simultaneously, many mitochondria in HT22 cells ruptured into punctiform instead of normal line shape after Hb exposure. Hence, we provided powerful evidence that mitochondrial injury including excessive fission and dysfunction occurred in the early stages of SAH.
Mitochondrial injury is associated with neuronal apoptosis and neurological deficits after SAH, and therefore, neurological scores, BBB permeability, and brain edema after SAH were deteriorated in our study. These results indicated that excessive mitochondrial fission may cause mitochondrial dysfunction and subsequent neuronal damage. Our study not only confirmed that mitochondrial damage occurred after SAH, in agreement with a previous study(Huang et al. 2013), but also showed the destruction of mitochondrial morphological homeostasis in neurons after SAH.
The mTOR pathway plays a key role in multiple cellular processes, including mRNA translation, ribosome biogenesis, autophagy, and metabolism (Ramanathan and Schreiber 2009). Previous studies showed that the mTOR pathway is involved in multiple types of stroke, and influences outcomes in animal models (Wang and Zhang 2017; Wang et al. 2018). We also confirmed that the activity of mTOR was closely correlated with neurological and mitochondrial function after SAH. Rapamycin, which inhibits mTOR by decreasing its phosphorylation (You et al. 2016), improved neurological outcomes and reduced BBB permeability and brain edema after SAH, as well the population of apoptotic cells in the cortex covered by blood clots. However, 3-MA administration exacerbated injury after SAH, which was in accordance with previous reports (Wang et al. 2012; Zhao et al. 2013). Current evidence indicates that mTOR regulates mitochondrial function in live cells (Morita et al. 2015). We, therefore, detected mTOR activity in neuronal mitochondrial function after SAH. ATP content, MMP, and ultrastructure were used to evaluate mitochondrial function. mTOR inhibition with rapamycin restored ATP levels in HT22 cells after Hb treatment, alleviated swelling of mitochondria and partially recovered mitochondrial ultrastructure in the SAH rats. The number of HT22 cells with depolarized MMP was also decreased in the Hb + rapamycin group, which indicated that less pro-apoptotic proteins were released from mitochondria into the cytoplasm (Mendelev et al. 2011). The quantitative analysis of Cyt C and cleaved caspase 3 in mitochondria and in the cytoplasm also confirmed the above findings. In contrast, the opposite findings were observed in the Hb + 3-MA group. A few cristae could still be found in the SAH group, but there were nearly no recognizable ultrastructures in the mitochondria of the SAH + 3-MA group. The ATP content and MMP in the cells of the Hb + 3-MA group were further decreased after the administration of 3-MA and pro-apoptotic proteins in the cytoplasm were upregulated. The cell viability and apoptosis rate of HT22 cells were consistent with the expression of apoptotic proteins in each group. Hence, we can conclude that inhibition of mTOR-improved neurological deficits and decreased neuronal injury and mitochondrial dysfunction after SAH, while activation of mTOR resulted in the opposite.
Mendl et al. reported that a decrease of MMP was accompanied by a change of mitochondrial dynamics and that the fusion was suppressed (Mendl et al. 2011). In addition, mitochondrial morphology is crucial for energy homeostasis, oxidative phosphorylation, reactive oxidative species management, calcium homeostasis and cell apoptosis in neurological diseases (Rose et al. 2017; An et al. 2019). Therefore, the function and morphology of mitochondria are inseparable. Rapamycin has been reported to affect mitochondrial changes in pathological states. Quesniaux reported rapamycin contribute to the continuous bone marrow cell proliferation by blocking the response to all major hematopoietic cytokines (Quesniaux 1993). A current study showed rapamycin can normalize mitochondrial size, number, and appearance via inhibition of mTOR complex 1 in the heart (Grevengoed et al. 2015). We thus speculated that mitochondrial morphology may also be influenced by mTOR activity as inhibition of mTOR-improved mitochondrial function after SAH. The final work of the present study was to detect alterations in mitochondrial morphology after administration of mTOR inhibitor or agonist in SAH models. Interestingly, super-resolution microscopy showed decreased mitochondrial length, circularity, and area after SAH, which was reversed after the administration of rapamycin, while 3-MA exacerbated excessive fission. The expression of Drp1 and p-Drp1 confirmed that excessive mitochondrial fission was improved after rapamycin treatment and was further exacerbated after 3-MA treatment. Importantly, we not only confirmed that excessive fission and mitochondrial dysfunction were the early events of neuronal apoptosis after SAH, but also found that inhibition of mTOR alleviated the excessive mitochondrial fission and dysfunction, which relieved the poor neuronal outcomes after SAH. In contrast, activation of mTOR further aggravated the mitochondrial and neuronal injury.
The present study provided evidence that the mTOR pathway is involved in neuroprotection against EBI after SAH via maintaining mitochondrial morphological homeostasis, and extends previous knowledge in regard to mTOR and regulation of autophagy. There are still some limitations in the study, rapamycin may also affect the mitochondrial changes via other pathways such as TNF-α (Quesniaux 1993). In addition, the mitochondrial fusion proteins Fis1 and Mfn were not detected, and that the mechanisms of mTOR on mitochondrial morphology need to be explored in future work.
Conclusion
Our present study demonstrated that the dynamic morphology of mitochondria in neurons appeared to be altered due to excessive fission after SAH, and that mitochondrial morphological homeostasis dysfunction was involved in neuronal injury and neurological deficits in EBI. However, inhibition of mTOR with rapamycin effectively decreased excessive mitochondrial fission, and alleviated mitochondrial and neuronal injury, while 3-MA further exacerbated these outcomes after SAH.
Change history
22 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10571-024-01477-2
Abbreviations
- BBB:
-
Blood–brain barrier
- CCA:
-
Common carotid artery
- Cyt C:
-
Cytochrome c
- DMSO:
-
Dimethylsulfoxide
- Drp1:
-
Dynamin-related protein 1
- EBI:
-
Early brain injury
- ECA:
-
External carotid artery
- GTP:
-
Guanosine-5′-triphosphate
- Hb:
-
Oxyhemoglobin
- ICA:
-
Internal carotid artery
- MMP:
-
Mitochondrial membrane potential
- mTOR:
-
Mammalian target of rapamycin
- PBS:
-
Phosphate-buffered saline
- ROS:
-
Reactive oxidative species
- SAH:
-
Subarachnoid hemorrhage
- TEM:
-
Transmission electron microscope
- 3-MA:
-
3-Methyl adenine
References
An J, Zhou Y, Zhang M, Xie Y, Ke S, Liu L, Pan X, Chen Z (2019) Exenatide alleviates mitochondrial dysfunction and cognitive impairment in the 5xFAD mouse model of Alzheimer’s disease. Behav Brain Res 370:111932
Cahill J, Calvert JW, Zhang JH (2006) Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 26:1341–1353
Carloni S, Buonocore G, Longini M, Proietti F, Balduini W (2012) Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience 203:160–169
Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, Molinari M, Viscomi MT, D’Amelio M (2014) Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death Dis 5:e1545
Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125:1241–1252
Cheng A, Hou Y, Mattson MP (2010) Mitochondria and neuroplasticity. ASN Neuro 2:e00045
Grevengoed TJ, Cooper DE, Young PA, Ellis JM, Coleman RA (2015) Loss of long-chain acyl-CoA synthetase isoform 1 impairs cardiac autophagy and mitochondrial structure through mechanistic target of rapamycin complex 1 activation. FASEB J 29:4641–4653
Guo F, Liu X, Cai H, Le W (2018) Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol 28:3–13
Hu P, Cheng B, He Y, Wei Z, Wu D, Meng Z (2018) Autophagy suppresses proliferation of HepG2 cells via inhibiting glypican-3/wnt/beta-catenin signaling. Onco Targets Ther 11:193–200
Huang L, Wan J, Chen Y, Wang Z, Hui L, Li Y, Xu D, Zhou W (2013) Inhibitory effects of p38 inhibitor against mitochondrial dysfunction in the early brain injury after subarachnoid hemorrhage in mice. Brain Res 1517:133–140
Ikeda Y, Sciarretta S, Nagarajan N, Rubattu S, Volpe M, Frati G, Sadoshima J (2014) New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev 2014:210934
Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G (2012) Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience 213:144–153
Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342:1524–1528
Kardideh B, Samimi Z, Norooznezhad F, Kiani S, Mansouri K (2019) Autophagy, cancer and angiogenesis: where is the link? Cell Biosci 9:65
Knott AB, Bossy-Wetzel E (2008) Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci 1147:283–292
Lee JY, He Y, Sagher O, Keep R, Hua Y, Xi G (2009) Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res 1287:126–135
Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J (2017) Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:1–19
Liu W, Tian F, Kurata T, Morimoto N, Abe K (2012) Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J Neurosci Res 90:1183–1189
Ma J, Meng F, Li S, Liu L, Zhao L, Liu Y, Hu Y, Li Z, Yao Y, Xi Z, Teng H, Xue Y (2015) Autophagy induction by endothelial-monocyte activating polypeptide II contributes to the inhibition of malignant biological behaviors by the combination of EMAP II with rapamycin in human glioblastoma. Front Mol Neurosci 8:74
Mendelev N, Mehta SL, Witherspoon S, He Q, Sexton JZ, Li PA (2011) Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion 11:76–82
Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS (2011) Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 124:1339–1350
Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I (2015) mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14:473–480
Pradeep H, Sharma B, Rajanikant GK (2014) Drp1 in ischemic neuronal death: an unusual suspect. Curr Med Chem 21:2183–2189
Quesniaux VF (1993) Immunosuppressants: tools to investigate the physiological role of cytokines. BioEssays 15:731–739
Ramanathan A, Schreiber SL (2009) Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci USA 106:22229–22232
Regmi SG, Rolland SG, Conradt B (2014) Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging 6:118–130
Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, Franco R (2017) Mitochondrial dysfunction in glial cells: implications for neuronal homeostasis and survival. Toxicology 391:109–115
Schuette AJ, Barrow DL (2013) Epidemiology and long-term mortality in subarachnoid hemorrhage. World Neurosurg 80:264–265
Shi ZY, Deng JX, Fu S, Wang L, Wang Q, Liu B, Li YQ, Deng JB (2017) Protective effect of autophagy in neural ischemia and hypoxia: negative regulation of the Wnt/beta-catenin pathway. Int J Mol Med 40:1699–1708
Sugawara T, Ayer R, Jadhav V, Zhang JH (2008) A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods 167:327–334
Sun Q, Wu W, Hu YC, Li H, Zhang D, Li S, Li W, Li WD, Ma B, Zhu JH, Zhou ML, Hang CH (2014) Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation 11:106
Sun XG, Duan H, Jing G, Wang G, Hou Y, Zhang M (2019) Inhibition of TREM-1 attenuates early brain injury after subarachnoid hemorrhage via downregulation of p38MAPK/MMP-9 and preservation of ZO-1. Neuroscience 406:369–375
Teng Z, Jiang L, Hu Q, He Y, Guo Z, Wu Y, Huang Z, Cao F, Cheng C, Sun X, Guo Z (2016) Peroxisome proliferator-activated receptor beta/delta alleviates early brain injury after subarachnoid hemorrhage in rats. Stroke 47:196–205
Tian Y, Li B, Shi WZ, Chang MZ, Zhang GJ, Di ZL, Liu Y (2014) Dynamin-related protein 1 inhibitors protect against ischemic toxicity through attenuating mitochondrial Ca2+ uptake from endoplasmic reticulum store in PC12 cells. Int J Mol Sci 15:3172–3185
Wang JP, Zhang MY (2017) Role for target of rapamycin (mTOR) signal pathway in regulating neuronal injury after intracerebral hemorrhage. Cell Physiol Biochem 41:145–153
Wang Z, Shi XY, Yin J, Zuo G, Zhang J, Chen G (2012) Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J Mol Neurosci 46:192–202
Wang P, Xie R, Cheng M, Sapolsky R, Ji X, Zhao H (2018) The mTOR cell signaling pathway is crucial to the long-term protective effects of ischemic postconditioning against stroke. Neurosci Lett 676:58–65
Wu P, Li Y, Zhu S, Wang C, Dai J, Zhang G, Zheng B, Xu S, Wang L, Zhang T, Zhou P, Zhang JH, Shi H (2017) Mdivi-1 alleviates early brain injury after experimental subarachnoid hemorrhage in rats, possibly via inhibition of Drp1-activated mitochondrial fission and oxidative stress. Neurochem Res 42:1449–1458
Xu T, Zhang WG, Sun J, Zhang Y, Lu JF, Han HB, Zhou CM, Yan JH (2015) Protective effects of thrombomodulin on microvascular permeability after subarachnoid hemorrhage in mouse model. Neuroscience 299:18–27
You W, Wang Z, Li H, Shen H, Xu X, Jia G, Chen G (2016) Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J Neurol Sci 367:224–231
Zhang N, Wang S, Li Y, Che L, Zhao Q (2013) A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett 535:104–109
Zhang T, Wu P, Zhang JH, Li Y, Xu S, Wang C, Wang L, Zhang G, Dai J, Zhu S, Liu Y, Liu B, Reis C, Shi H (2018) Docosahexaenoic acid alleviates oxidative stress-based apoptosis via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell Mol Neurobiol 38:1413–1423
Zhao H, Ji Z, Tang D, Yan C, Zhao W, Gao C (2013) Role of autophagy in early brain injury after subarachnoid hemorrhage in rats. Mol Biol Rep 40:819–827
Zhao H, Li Y, Chen L, Shen C, Xiao Z, Xu R, Wang J, Luo Y (2019) HucMSCs-derived miR-206-knockdown exosomes contribute to neuroprotection in subarachnoid hemorrhage induced early brain injury by targeting BDNF. Neuroscience 417:11–23
Acknowledgements
This work is supported by Natural Science Foundation of Heilongjiang Province, China (ZD2018018) and Innovation Fund of Harbin Medical University, China (YTSKYCX2018-38HYD).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Fig. 4A has been corrected.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Wu, P., Dai, J. et al. Inhibition of mTOR Alleviates Early Brain Injury After Subarachnoid Hemorrhage Via Relieving Excessive Mitochondrial Fission. Cell Mol Neurobiol 40, 629–642 (2020). https://doi.org/10.1007/s10571-019-00760-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00760-x