
Abstract
Autophagy is a self-degradative process and it plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, and eliminating intracellular pathogens. Previous studies have demonstrated that autophagy pathway was activated in brain after experimental subarachnoid hemorrhage (SAH); however, the role of autophagy in the pathogenesis of early brain injury (EBI) following SAH remains unknown. Experiment 1 aimed to investigate the time–course of the autophagy in the cortex following SAH. In experiment 2, we chose the maximum time pointof autophagy activation and assessed the effects of rapamycin (RAP, autophagy activator) and 3-methyladenine (3-MA, autophagy inhibitor) on regulation of EBI. All SAH animals were subjected to injection of 0.3 ml fresh arterial, nonheparinized blood into prechiasmatic cistern in 20 s. As a result, microtubule-associated protein light chain-3 (LC3), a biomarker of autophagosome, and beclin-1, a Bcl-2-interacting protein required for autophagy, were significantly increased at the early stage of SAH and their expressions peaked at 24 h after SAH. In RAP-treated group, the early brain damage such as brain edema, blood–brain barrier (BBB) impairment, cortical apoptosis, and clinical behavior scale was significantly ameliorated in comparison with vehicle-treated SAH rats. Conversely, 3-MA decreased expression of LC3 and beclin-1, increased the average value of brain edema and BBB disfunction, and aggravated neurological deficits. Our results suggest that autophagy pathway is activated in the brain after SAH and may play a beneficial role to EBI development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spontaneous subarachnoid hemorrhage (SAH) is a devastating disease and carrying a mortality rate ranging from 32% to 67% (Hop et al. 1997). In the past decades, vasospasm has long been considered the most dreaded complication after SAH and a lot of work has concentrated primarily on it. But the high morbidity and mortality associated with SAH have not changed despite major advances in surgical techniques, radiology, and anesthesiology. Furthermore, research has pointed out that early brain injury (EBI) is the primary cause of mortality in SAH patients (Broderick et al. 1994). Accumulated evidence suggests that the primary cause of poor outcome after SAH is not only vasospasm but also EBI. Therefore, Treatment of EBI has been considered as a major goal in the management of patients surviving SAH. However, the exact molecular mechanism of EBI still remains obscure, which has hindered the development of effective and specific treatment paradigms for EBI. The term EBI refers to the immediate injury to the brain as a whole, within the first 48 h of the ictus, secondary to SAH (Ostrowski et al. 2006). The possible mechanisms about EBI may include: elevation of intracranial pressure (ICP), reduction of cerebral blood flow (CBF), suppression of cerebral perfusion pressure (CPP), fall in brain oxygenation, blood–brain barrier (BBB) breakdown, brain edema, and neuronal cell death (Cahill and Zhang 2009).
Autophagy is a self-degradative process that is important for balancing sources of energy at critical times in development and in response to nutrient stress. Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, and eliminating intracellular pathogens (Glick et al. 2010). Autophagy pathway has been reported to play an important role in several central nervous system (CNS) diseases such as cerebral ischemia (Liu et al. 2010), hypoxia–ischemia-induced brain injury (Carloni et al. 2008), and traumatic brain injury (Zhang et al. 2008). The common features of these CNS disorders also included increased ICP, decreased CBF, insufficient CPP, brain edema, and BBB dysfunction, which were in accordance with the pathophysiology of EBI following SAH.
A previous report from Lee et al. (2009) has indicated that autophagy was significantly increased in the cortex immediately following SAH and last during the entire phase of early brain injury. However until now, no study was found in the literature to investigate the potential contribution of autophagy to SAH-induced EBI and also little work has been done concerning the effects of rapamycin (RAP, autophagy activator) and 3-methyladenine (3-MA, autophagy inhibitor) on brain damage during the early stage of SAH. The aim of the current study was to evaluate the cortical activity of autophagy pathway following SAH and determine the potential role of autophagy in the development of EBI. We hypothesized that the activating autophagy might attenuate the development of EBI in this rat SAH model.
Materials and Methods
Animals
The animal use and care protocols, including all operation procedures, were approved by the Animal Care and Use Committee of Soochow University and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institute of Health. One hundred and forty-three male Sprague–Dawley rats weighing from 280 to 325 g were purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). They were acclimated in a humidified room and maintained on the standard pellet diet at the Animal Center of Soochow University for 10 days before the experiment. The temperature in both the feeding room and the operation room was maintained at about 25°C.
Prechiasmatic Cistern SAH Model
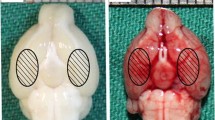
Following intraperitoneal anesthesia with 4% chloral hydrate (400 mg/kg body weight), animal head was fixed in the stereotactic frame. Experimental SAH model was produced using stereotaxic insertion of a needle with a rounded tip and a side hole into the prechiasmatic cistern according to our previous study (Wang et al. 2010). The needle was tilted 45° in the sagittal plane, and placed 7.5 mm anterior to bregma in the midline, with the hole facing the right side. It was lowered until the tip reached the base of the skull, 2–3 mm anterior to the chiasma (about 10–12 mm from the brain surface), and retracted 0.5 mm. Loss of cerebrospinal fluid and bleeding from the midline vessels were prevented by plugging the burr hole with bone wax before inserting the needle. The amount of 0.3 ml nonheparinized fresh autologous arterial blood was slowly injected into the prechiasmatic cistern for 20 s under aseptic technique. Control animals were injected with 0.3 ml saline. The animals were allowed to recover 45 min after SAH. After operation procedures, the rats were then returned to their cages and the room temperature kept at 23 ± 1°C. 20 ml of 0.9% NaCl was injected subcutaneously right after the operation to prevent dehydration. Heart rate and rectal temperature were monitored and the rectal temperature was kept at 37 ± 0.5°C, by using physical cooling (ice bag) when required, throughout experiments. It was observed in the present study that inferior basal temporal lobe was always stained by blood. Hence, the brain tissue adjacent to the clotted blood was taken to analysis in our study (Fig. 1). Control animals underwent exactly the same procedure as described above with the exception that no blood was injected intracisternally.

Schematic representation of the areas taken for assay. a Control rat brain, b SAH rat brain, c H&E staining of control rat brain, d H&E staining of SAH rat brain
Experimental Design
In experiment 1, 25 rats were assigned randomly to five groups: control group, SAH 6-, 12-, 24-, and 48-h groups. The animals in SAH 6-, 12-, 24-, and 48-h groups were subjected to experimental SAH and were killed at 6, 12, 24, and 48 h after blood injection, respectively (n = 5 for each group).
The groups in experiment 2 consisted of control group (n = 15), SAH group (n = 15), SAH + vehicle group (n = 15), SAH + RAP group (n = 15), and SAH + 3-MA group (n = 15). RAP and 3-MA (Sigma-Aldrich Inc., St. Louis, MO, USA) were dissolved in methyl sulfoxide (DMSO, Sigma-Aldrich Inc.) and the proportion was 3.3 mol/L (RAP) and 0.1 mol/L (3-MA), respectively. All the rats in experiment 2 were killed at 24 h post-SAH.
Drug Administration
The method of intracerebroventricular infusion was performed according to a previous study (Smith et al. 1995). The needle of a 5-μL syringe (Shanghai Gaoge Industry & Trade Co. Ltd) was inserted through a burr hole perforated on the skull into the right lateral ventricle using the following coordinates relative to bregma: 1.5 mm posterior, 1.0 mm lateral, and 3.2 mm below the horizontal plane of bregma. DMSO vehicle (3 μL), RAP (3 μL), or 3-MA (3 μL) was infused at 20 min before the blood injection. The needle was removed after finishing infusion and the burr hole was quickly plugged with a bone wax. The dose was chosen according to previous reports (Wen et al. 2008; Zhang et al. 2009) since they observed beneficial effects in models of cerebral ischemia and Alzheimer’s disease using the same dose.
Immunohistochemical Study
Immunohistochemistry on formalin-fixed paraffin-embedded sections was performed to determine the immunoreactivity of microtubule-associated protein light chain-3 (LC3), beclin-1, and Ig G. Sections were deparaffinized and rehydrated in graded concentrations of ethanol to distilled water. Endogenous peroxidase activity was blocked with 3% H2O2 for 5 min followed by a brief rinse in distilled water and a 15-min wash in PBS. Sections were placed in 10 mmol/L citrate buffer (pH 6.0), and heated in microwave oven at 95°C for 30 min. Sections were cooled at room temperature for 20 min and rinsed in PBS. Non-specific protein binding was blocked by 40 min incubation in 5% horse serum. Sections were incubated with primary antibodies (anti-LC3, anti-beclin-3 and anti-Ig G, all diluted 1:200, respectively, from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h at room temperature, followed by a 15-min wash in PBS. Sections were incubated with horseradish peroxidase (HRP)-conjugated IgG (1:500 dilution, Santa Cruz Biotechnology, Inc.) for 60 min at room temperature. DAB was used as chromogen and counterstaining was done with hematoxylin. Sections incubated in the absence of primary antibody were used as negative controls. Microscopy of the immunohistochemically stained tissue sections was performed by an experienced pathologist blinded to the experimental condition. Evaluation of sections was undertaken by assessing the intensity of staining (four grades); “1” indicates very low density of positive cells, “2” indicates a moderate density of positive cells, “3” indicates the higher, but not maximal density of positive cells, and “4” indicates the highest density of positive cells.
Western Blot Analysis
The frozen brain samples were mechanically lysed in 20 mM Tris, pH 7.6, which contains 0.2% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, 1 mM phenylmethylsulphonyl fluoride, and 0.11 IU/ml aprotinin (all purchased from Sigma-Aldrich, Inc.). Lysates were centrifuged at 12,000×g for 20 min at 4°C. The protein concentration was estimated by the Bradford method using the Nanjing Jiancheng protein assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The samples (60 μg per lane) were separated by 8% SDS-polyacrylamide gel electrophoresis and electro-transferred onto a polyvinylidene-difluoride membrane (Bio-Rad Lab, Hercules, CA, USA). The membrane was blocked with 5% skimmed milk for 2 h at room temperature, incubated overnight at 4°C with primary antibodies directed against LC-3 and beclin-1 (both from Santa Cruz Biotechnology) at the dilutions of 1:200 and 1:150, respectively. The glyceraldehyde-3-phosphate dehydrogenase (diluted in 1:6000, Sigma-Aldrich, Inc.) was used as a loading control. After the membrane was washed for 10 min each for six times in PBS + Tween 20 (PBST), it was incubated in the appropriate HRP-conjugated secondary antibody (diluted 1:400 in PBST) for 2 h. The blotted protein bands were visualized by enhanced chemiluminescence Western blot detection reagents (Amersham, Arlington Heights, IL, USA) and were exposed to X-ray film. Developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp, Nagano, Japan). Optical densities were obtained using Glyko Bandscan software (Glyko, Novato, CA, USA). The tissue of six animals was used for Western blot analysis at each time point. All experiments have been repeated at least three times.
Brain Water Content
Brain edema was determined using the wet/dry method as previously described where % brain water = [(wet weight − dry weight)/wet weight] × 100% (Turkoglu et al. 2008). Briefly, brain samples were rapidly removed from the skull and placed separately into preweighed and labeled glass vials and weighed. The vials were then placed in an oven for 72 h at 100°C and then reweighed to obtain dry weight content.
Blood–Brain Barrier Permeability
BBB permeability was determined by Evans blue (EB) extravasation at 24 h after SAH (Kohta et al. 2007). Briefly, 2% Evans blue was injected intravenously at a dose of 2 ml/kg. Animals were then re-anesthetized after 1 h with urethane (1,000 mg/kg) and perfused using saline to remove intravascular EB dye. Animals were then decapitated, the brains removed and homogenized in phosphate-buffered saline. Trichloroacetic acid was then added to precipitate protein, and the samples were cooled and centrifuged. The resulting supernatant was measured for absorbance of EB at 610 nm using a spectrophotometer.
TUNEL Staining and Quantitation of Apoptotic Cells
The formalin-fixed tissues were embedded in paraffin and sectioned at 4 μm thickness with a microtome. The sections were detected for apoptotic cells by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) method. TUNEL: In situ cell death detection Kit POD (ISCDD, Boehringer Mannheim, Germany) was used. The procedures were according to protocol of the kit. The positive cells were identified, counted, and analyzed under the light microscope by an investigator blinded to the grouping. The extent of brain damage was evaluated by apoptotic index which was the average number of TUNEL-positive cells in each section counted in 10 microscopic fields (at ×200 magnifications).
Neurologic Scoring
Three behavioral activity examinations (Table 1) were performed at 24 h after SAH using the scoring system reported previously to record appetite, activity and neurological deficits (Yamaguchi et al. 2004).
Statistical Analysis
All data were presented as mean ± SD. SPSS 12.0 was used for statistical analysis of the data. The Mann–Whitney U test was used to compare the behavior and activity score among groups. The other measurements were analyzed by one-way analysis of variance or Student’s t test. Statistical significance was inferred at P < 0.05.
Results
General Observation
There were no significant differences in body weight, temperature, or injected arterial blood gas data among the experimental groups (data not shown). After induction of SAH, all animals will stop breathing for about 30 s. The mortality rate of rats in the control group was 0% (0/20 rats), and it was 35% (43/123 rats) in the SAH group.
Western Blot Analysis for Detecting Autophagy Activation after SAH
Western blot analysis showed low level of LC3 and beclin-1 in the control group (Fig. 2). The expressions of LC3 and beclin-1 was significantly increased after SAH and peaked at 24 h after blood injection (Fig. 2). For beclin-1, there is no statistically significant difference between SAH 48-h group and control group (Fig. 2).

Upper representative autoradiogram of LC3 and beclin-1 in the cortex. We observed LC-3 at 16 kDa, beclin-1 at 52 kDa and a loading control glyceraldehyde-3-phosphate dehydrogenase at 37 kDa. It shows that the expression of autophagy-related proteins increased after SAH and peaked at 24 h following SAH. Bottom quantitative analysis of the Western blot results for LC3 and beclin-1. It shows that the levels of LC3 and beclin-1 in SAH 6-, 12-, and 24-h groups are significantly higher than that in control. Bars represent the mean ± SD (n = 5, each group). *P < 0.05 compared with control group, **P < 0.01 compared with control group, # P < 0.05 compared with SAH 12- or 48-h groups, ns P > 0.05 compared with control group
Immunohistochemistry for LC3 and Beclin-1 Expression after SAH
To assess the localization of autophagy activation, immunohistochemistry for LC3 and beclin-1 was performed. A few LC3 or beclin-1 positive cells were observed in the control group, which indicates the constitutional activity of autophagy in the normal brain of rats (Fig. 3a, f, and Fig. 4). Increased LC3 or beclin-1 positive cells in the SAH groups could be found in the brain samples (Fig. 3b–d, g–i). LC3 and beclin-1 immunoreactivity was mainly present in the neurons (Fig. 3d and i). Quantitative analysis showed the low LC3 and beclin-1 immunoreactivity in the control group with the average scores of 1.24 ± 0.17 and 1.36 ± 0.09, respectively (Fig. 4). In contrast, autophagy was activated at SAH 6-, 12-, and 24-h groups (Fig. 4). Significant differences were both found between the SAH 6-, 12-, and 24-h groups, and the control group (P < 0.05, Fig. 4). Autophagy was recovered to the baseline in this SAH model and there was no significant difference between SAH 48-h group and control group (P > 0.05, Fig. 4).

Immunohistochemical study of LC3 and beclin-1 on brain samples (scale bar 100 μm). a and f Almost undetectable LC3 and beclin-1 expression in the cortex from control rats; b and g moderate LC3 and beclin-1 immunoreactivity in the neurons at 6 h post-SAH; c and h strong LC3 and beclin-1 immunoreactivity in the brain tissue at 12 h after blood injection; d and i very strong LC3 and beclin-1 immunoreactivity in the neurons of SAH brains at SAH 24-h group (arrows show positive cells); e and j low expression of LC3 and beclin-1 was shown in SAH 48-h group

Quantitative analysis showed the low expression of LC3 and beclin-1 in the control group. In contrast, the level of autophagy was increased in SAH 6-, 12-, and 24-h groups. There is no statistically significant difference between SAH 48-h group and control group. Bars represent the mean ± SD (n = 5, each group). **P < 0.01 compared with control group, *P < 0.05 compared with control group, ns P > 0.05 compared with control group
Effect of Autophagy Activator (RAP) or Inhibitor (3-MA) on LC3 and Beclin-1 Expression in SAH Brains
To determine the influence of RAP or 3-MA on autophagy activation in the cortex post SAH, western blot analysis was performed to detect the changes of LC3 and beclin-1 as described in “Materials and Methods” section. Figure 5 showed low level of LC3 and beclin-1 expression in control group. At 24 h after SAH, the levels of LC3 and beclin-1 were significantly increased in SAH group and SAH + vehicle group (P < 0.01; Fig. 5). There was no statistically significant difference between SAH group and SAH + vehicle group (P > 0.05; Fig. 5). After RAP injection, the level of LC3 and beclin-1 was markedly upregulated in animals of SAH + RAP group (P < 0.05; Fig. 5). Conversely, after 3-MA injection, the increased activity of autophagy was markedly suppressed in animals of SAH + 3-MA group (P < 0.01; Fig. 5).

Expressions of LC3 and beclin-1 in the brains in control group (n = 5), SAH group (n = 5), SAH + vehicle group (n = 5), SAH + RAP group (n = 5), and SAH + 3-MA group (n = 5). Upper representative autoradiogram of the proteins expression following SAH by western blot. We detected LC3 at 16 kDa, beclin-1 at 52 kDa, and the loading control glyceraldehyde-3-phosphate dehydrogenase at 37 kD. 1 and 5 control groups, 2 and 6 SAH groups, 3 and 7 SAH + vehicle groups, 4 SAH + RAP group, 8 SAH + 3-MA group. Bottom Quantitative analysis of the western blot results for the levels of LC3 and beclin-1. The activity of autophagy was significantly increased in the animals of SAH and SAH + vehicle groups compared with the control group (P < 0.01). The increased activity was further markedly upregulated by RAP treatment (P < 0.05) and suppressed by 3-MA administration (P < 0.01). Bars represent the mean ± SD. **P < 0.01 compared with control group, ns P > 0.05 compared with SAH group, # P < 0.05 compared with SAH + vehicle group, ## P < 0.01 compared with SAH + vehicle group
Autophagy Activator (RAP) Administration Improved Blood–Brain Barrier Permeability after SAH
The pattern of Evans blue extravasation following SAH is shown in Table 2. Rats in SAH group demonstrated a significant increase (P < 0.01) in BBB permeability to Evans blue relative to rats of control group. Administration of RAP significantly inhibited Evans blue extravasation (P < 0.01), indicating a reduced BBB opening in response to autophagy upregulation. However, no significant difference was seen between the SAH + 3-MA group and the SAH + vehicle group (P = 0.064).
IgG Immunoreactivity Changed in Autophagy Activator (RAP) or Inhibitor (3-MA)-Treated SAH Brains
Low IgG immunoreactivity was found in the control group rat brain samples (Fig. 6a). In SAH and SAH + vehicle groups, the IgG expression in cerebral cortex was found to be significantly increased compared with those in control animals (P < 0.01; Fig. 6b and c). In SAH + 3-MA group, when compared with that in the SAH + vehicle group, the IgG extravasation in the studied brain tissue was significantly increased (P < 0.05; Fig. 6d). In RAP-treated rats, IgG expression was significantly downregulated as compared with vehicle-treated group (P < 0.01; Fig. 6e). The result showed that activating autophagy following SAH could attenuate BBB dysfunction.

IgG immunohistochemistry staining of the brains in control group (n = 5), SAH group (n = 5), SAH + vehicle group (n = 5), SAH + RAP group (n = 5), and SAH + 3-MA group (n = 5). a Control group rats showing low IgG staining; (b) SAH group rats showing increased IgG expression stained brown (arrows show IgG extravasation). c In SAH + vehicle group, it can also be seen IgG expression in the cortex. d SAH + 3-MA group rats showing stronger IgG staining than SAH + vehicle group. e In SAH + RAP group, low IgG protein expression was shown. f Administration of 3-MA or RAP significantly regulated the IgG expression in studied cortex following SAH. Bars represent the mean ± SD. **P < 0.01 compared with control group, ns P > 0.05 compared with SAH group, ## P < 0.01 compared with SAH + vehicle group, # P < 0.05 compared with SAH + vehicle group
RAP Administration Alleviated Brain Edema after SAH
Significant increase (P < 0.01) in water content was detected in the brain samples at 24 h after SAH when compared with control-operated rats (Fig. 7). The mean value of brain water content was decreased by RAP administration (P < 0.01) as compared with SAH + vehicle group. For 3-MA administration, the mean values of brain water content had no significant difference between SAH + 3-MA group and vehicle-treated group (P > 0.05). The results suggested that post-SAH RAP treatment could attenuate brain edema in this rat SAH model.

Alterations in brain water content in control group (n = 5), SAH group (n = 5), SAH + vehicle group (n = 5), SAH + RAP group (n = 5), and SAH + 3-MA group (n = 5). The brain water content was increased significantly at 24 h after SAH. RAP treatment markedly reduced brain water content. No difference of brain water content was detected between SAH + 3-MA group and SAH + vehicle group. **P < 0.01 compared with control group, ns P > 0.05 compared with SAH group, ## P < 0.01 compared with SAH + vehicle group, † P = 0.058 compared with SAH + vehicle group
RAP Administration Repressed Cortical Apoptosis in the Brain after SAH
Few TUNEL-positive apoptotic cells were found in the control group rat brains (Fig. 8a). In SAH and SAH + vehicle groups, the apoptotic index in the cortex was found to be significantly increased compared with those in control animals (P < 0.01; Fig. 8b and c). There was no statistically significant difference between SAH group and SAH + vehicle group (P > 0.05). In SAH + RAP group, when compared with that in the SAH + vehicle group, the apoptotic index in the studied cortex was significantly decreased (P < 0.05, Fig. 8d), but in SAH + 3-MA group, the index was increased remarkably (P < 0.05, Fig. 8e).

TUNEL immunohistochemistry staining of the cortex in control group (n = 5), SAH group (n = 5), SAH + vehicle group (n = 5), SAH + RAP group (n = 5), and SAH + 3-MA group (n = 5). a Control group rats showing few TUNEL apoptotic cells, b SAH group rats showing strong TUNEL staining, c SAH + vehicle group rats showing more TUNEL apoptotic cells stained brown (arrows show positive cells), d SAH + 3-MA group rats showing more TUNEL apoptotic cells than SAH or SAH + vehicle group, e SAH + RAP group rats showing less TUNEL apoptotic cells than SAH or SAH + vehicle group (scale bar 100 μm). f Administration of RAP significantly decreased the apoptotic index in rat brain following SAH. **P < 0.01 versus control group, # P < 0.05 versus SAH + vehicle group, ns P > 0.05 versus SAH group
Impact of Autophagy Activator (RAP) or Inhibitor (3-MA) on Clinical Behavior Function after Experimental SAH
As compared with control group, clinical behavior function impairment caused by SAH was evident in SAH subjects (P < 0.01, Table 3). No significant difference was seen between the SAH group and SAH + vehicle group (P > 0.05). RAP-treated rats showed better performance in this scale system than vehicle-treated rats at 24 h after SAH (Table 3), and the difference was statistically significant (P < 0.01, Table 3). After 3-MA administration, the animals showed worse clinical symptoms as compared with vehicle-treated rats (P < 0.05, Table 3).
Discussion
The main findings of this study are as follows: (1) autophagy was activated in the brain during early stage after SAH in rats, which was in agreement with the previous study (Lee et al. 2009); (2) the time–course study showed the autophagy peaked at 24 h and recovered at 48 h following SAH; (3) SAH-induced increases of LC3 and beclin-1 could be significantly modulated by intracerebroventricular administration of RAP or 3-MA, which proved that autophagy was regulated by the treatments in this SAH model; (4) the activator of autophagy, RAP, could ameliorate the brain edema, BBB permeability, and clinical behavior function; (5) after administration of autophagy inhibitor (3-MA), the parameters concerning EBI were remarkably aggravated in this prechiasmatic blood injection model. These findings suggest for the first time that activation of autophagy pathway may play a potential role to attenuate the development of EBI in the rat SAH model.
Accumulating evidence has demonstrated that the autophagy pathway plays important roles in the pathogenesis of different diseases in the CNS, such as cerebral ischemia (Liu et al. 2010), traumatic brain injury (Zhang et al. 2008), experimental intracerebral hemorrhage (He et al. 2008), and hypoxia-ischemia brain injury (Carloni et al. 2008). Till now, no study was found in the literature to investigate the role of autophagy in EBI after SAH. In brain trauma field, Zhang et al. (2008) investigated changes of autophagy after traumatic brain injury (TBI) and its possible role by using controlled cortical injury model. The authors demonstrated that autophagy was activated and might protect neurons from degeneration at early stage post-TBI and play a continuous role afterwards in eliminating aberrant cell components. In the hypoxia–ischemia-induced brain injury model, previous report (Carloni et al. 2008) indicated that activation of autophagy may be a component of the survival mechanisms turned on after brain ischemia and limiting over-activation autophagic pathways could delay the progression of cells towards death, probably by providing cells with metabolic substrates to meet their energetic demands and sustaining survival programs.
The only study about autophagy and SAH was from Lee et al. (2009), which demonstrated that a significantly increased autophagic activity in the cortex in the phase of EBI after SAH, but the role of autophagy in SAH-induced EBI was not mentioned in the publication. In their research, intracranial endovascular perforation SAH model was used, which provided little control over the amount of blood released in the subarachnoid space, resulting in an experimental model with variable bleeding. At the same time, the mortality was almost 50% after inducing SAH. In the current research, we produced SAH by prechiasmatic blood injection model, which has been proved to be the most suitable for studies of the pathological and pathophysiological processes of SAH after comparison with intracranial endovascular perforation SAH model and cisterna magna SAH model. The prechiasmatic SAH model could induce a significant acute reduction in CBF, combined with a substantial degree of neuronal death. The mortality rate is acceptable, and the procedure is reproducible and easy to perform (Prunell et al. 2004). At the same time, present data concerning EBI showed higher edema index, evens blue value, IgG extravasation, and worse clinical score compared with control rats, which meant that the model is an appropriate one for EBI research.
Brain edema represents a major component of EBI following SAH as a direct consequence of the BBB dysfunction (Kusaka et al. 2004) and not as a result of vasospasm (Claassen et al. 2002). Although brain edema secondary to SAH has been largely ignored in the literature, Classen et al. showed that 8% of patients had global cerebral edema detected by CT scan on admission and that an additional 12% developed appreciable edema over the first 6 days (Claassen et al. 2002; Kassell et al. 1990). In this present study, our data demonstrated that a significant increase in water content and BBB dysfunction was found in the brain 24 h following SAH induction, which could be alleviated by autophagy activator administration.
Autophagy is a major cellular pathway for the degradation of long-lived proteins and cytoplasmic organelles in eukaryotic cells (Cuervo 2004). A large number of intracellular/extracellular stimuli, including amino acid starvation and invasion of microorganisms, are able to induce the autophagic response. Under pathological conditions, autophagy and apoptosis interacts each other to determine cells’ fate (Kegel et al. 2000). In cells, autophagy both serves as a defense mechanism to resist cell injuries caused by circumstance changes and induces cell death that differs from apoptosis (Bursch et al. 2000). Autophagy has either deleterious or protective effects depending on the specific situation and stage in the pathological process (Rubinsztein et al. 2005). Many pathological conditions, including cancer and neurodegenerative disorders, are associated with autophagy (Shintani and Klionsky 2004).
Rapamycin, a macrolide antibiotic, is a widely used inhibitor of the mammalian target of rapamycin which induces autophagy in a variety of in vivo or vitro models. The protein target of rapamycin (Tor) plays a major regulatory role in autophagy induction. Tor forms two functionally distinct protein complexes, Tor complex 1 and 2 (TORC1 and TORC2), and TORC1 has the primary role in regulating autophagy. Under nutrient-rich conditions, TORC1 is active and inhibits autophagy, whereas TORC1 is inhibited upon nutrient deprivation, allowing an increase in autophagic activity (Rangaraju et al. 2010). As mentioned by Nührenberg et al. (2005), besides the modulating effect of autophagy pathway, rapamycin has profound inhibitory influence on the proinflammatory gene expression pattern and on promoters of hematopoetic progenitor cells (HPC) after vascular injury. In their study, the data demonstrated that rapamycin suppressed the coordinated proadhesive and proinflammatory gene expression pattern next to downregulation of genes related to metabolism, proliferation, and apoptosis and could lead to a reduced recruitment of leukocytes and HPC after vascular injury. However nowadays, rapamycin administration model has been a classical one for autophagy research (Sarkar et al. 2009; Ravikumar et al. 2004).Our results indicated that SAH activated autophagy, and the activation may begin at 6 h or earlier, and last at least 24 h after SAH. Autophagy may play a continuous role in the bulk elimination of aberrant cell components afterwards, which contributes to the cellular homeostasis of the brain. The number of LC3(+) and beclin-1(+) cells peaked at 24 h following SAH, suggesting that autophagy played an important role in EBI following SAH, because autophagy was involved in elimination of impaired cells in a non-apoptotic cell death pathway.
In summary, to the best of our knowledge, this present study is the first one to demonstrate the protective contribution of autophagy to EBI in the experimental SAH model, which suggests that autophagy activation and signaling could participate in the pathogenesis of EBI induced by SAH. Activation of autophagy in this SAH model resulted in reduction of brain edema and the degree of BBB disruption following SAH. Moreover, the clinical behavior scales were improved by administration of autophagy activator (RAP). So autophagy may play a potential role for alleviating the EBI, which will provide novel ideas for pursuing therapeutic agents for SAH-induced EBI.
References
Hop JW, Rinkel GJ, Algra A, van Gijn J (1997) Case-fatality rates and functional outcome after subarachnoid hemorrhage: a systematic review. Stroke 28:660–664
Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A (1994) Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 25:1342–1347
Ostrowski RP, Colohan AR, Zhang JH (2006) Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res 28:399–414
Cahill J, Zhang JH (2009) Subarachnoid hemorrhage: is it time for a new direction? Stroke 40:S86–S87
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221:3–12
Liu C, Gao Y, Barrett J, Hu B (2010) Autophagy and protein aggregation after brain ischemia. J Neurochem 115:68–78
Carloni S, Buonocore G, Balduini W (2008) Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 32:329–339
Zhang YB, Li SX, Chen XP, Yang L, Zhang YG, Liu R et al (2008) Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull 24:143–149
Lee JY, He Y, Sagher O, Keep R, Hua Y, Xi G (2009) Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res 1287:126–135
Wang Z, Chen G, Zhu WW, Bian JY, Shen XO, Zhou D (2010) Influence of simvastatin on microthrombosis in the brain after subarachnoid hemorrhage in rats: a preliminary study. Ann Clin Lab Sci 40:32–42
Smith RA, Balis FM, Ott KH, Elsberry DD, Sherman MR, Saifer MG (1995) Pharmacokinetics and tolerability of ventricularly administered superoxide dismutase in monkeys and preliminary clinical observations in familial ALS. J Neurol Sci 129:13–18
Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD et al (2008) Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4:762–769
Zhang JY, Peng C, Shi H, Wang S, Wang Q, Wang JZ (2009) Inhibition of autophagy causes tau proteolysis by activating calpain in rat brain. J Alzheimers Dis 16:39–47
Turkoglu OF, Eroglu H, Okutan O, Tun MK, Bodur E, Sargon MF, Oner L, Beskonakli E (2008) A comparative study of treatment for brain edema: magnesium sulphate versus dexamethasone sodium phosphate. J Clin Neurosci 15:60–65
Kohta M, Minami H, Tanaka K, Kuwamura K, Kondoh T, Kohmura E (2007) Delayed onset massive oedema and deterioration in traumatic brain injury. J Clin Neurosci 14:167–170
Yamaguchi M, Zhou C, Nanda A, Zhang JH (2004) Ras protein contributes to cerebral vasospasm in a canine double-hemorrhage model. Stroke 35:1750–1755
He Y, Wan S, Hua Y, Keep RF, Xi G (2008) Autophagy after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab 28:897–905
Prunell GF, Mathiesen T, Svendgaard NA (2004) Experimental subarachnoid hemorrhage: cerebral blood flow and brain metabolism during the acute phase in three different models in the rat. Neurosurgery 54:426–436
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH (2004) Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 24:916–925
Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA (2002) Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke 33:1225–1232
Kassell NF, Torner JC, Haley EC, Jane JA, Adams HP, Kongable GL (1990) The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 73:18–36
Cuervo AM (2004) Autophagy: in sickness and in health. Trends Cell Biol 14:70–77
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N et al (2000) Huntingtin expression stimulates endosomal–lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20:7268–7278
Bursch W, Ellinger A, Gerner C, Fröhwein U, Schulte-Hermann R (2000) Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci 926:1–12
Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B et al (2005) Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1:11–22
Shintani T, Klionsky DJ (2004) Autophgy in health and disease: a doubleedged sword. Science 306:990–995
Rangaraju S, Verrier JD, Madorsky I, Nicks J, Dunn WA Jr, Notterpek L (2010) Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci 30:11388–11397
Nührenberg TG, Voisard R, Fahlisch F, Rudelius M, Braun J, Gschwend J et al (2005) Rapamycin attenuates vascular wall inflammation and progenitor cell promoters after angioplasty. FASEB J 19:246–248
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595
Acknowledgments
This work was supported by the 135 grant from Health Department of Jiangsu Province (RC2007081) and a grant from the China Postdoctoral Science Foundation (No.20090460085).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhong Wang and Xiao-Yong Shi contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, Z., Shi, XY., Yin, J. et al. Role of Autophagy in Early Brain Injury after Experimental Subarachnoid Hemorrhage. J Mol Neurosci 46, 192–202 (2012). https://doi.org/10.1007/s12031-011-9575-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9575-6