
Abstract
Noncovalent interactions are vitally important to understand the structural stability and molecular assembly of cellulose and its analog molecules. Using density functional theory in conjunction with three popular generations of dispersion correction (D2, D3, D4), we systematically estimate the strength of inter-chain interaction for several β-1,4-linked crystalline polysaccharides (cellulose Iα, Iβ, II, IIII, α-chitin, β-chitin, chitosan) and their building block monomers (glucose, cellobiose). Switching on and off dispersion correction and combining the calculation of condensed and isolated chains allow the extraction of the intra- and inter-chain London dispersion interactions and the inter-chain electrostatic interaction. Regardless of the generations of dispersion correction and allomorphs, the estimated inter-chain London dispersion interaction is 45 ~ 74 kJ/mol per pyranose ring comparable to the inter-chain electrostatic interaction (47 ~ 88 kJ/mol). The upper limit of the strength of inter- or intra-chain hydrogen bonds is estimated to be 27 ~ 50 or 21 ~ 53 kJ/mol, respectively, based on energy profiles of hydroxy rotation. Our work quantitatively highlights that it is the London dispersion interaction rather than the hydrogen bonding interaction dominating in the tight assembly of polymer chains for β-1,4-linked crystalline polysaccharides, regardless of the crystal allomorph and types as well as the generations of dispersion correction of DFT. Thus, London dispersion interaction should be preferentially considered during their deconstruction, defibrillation, or dissolution processes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
X-ray and neutron studies revealed abundant hydrogen bonds and regular network patterns among the crystal structures of cellulose and chitin (Langan et al. 1999; Nishiyama et al. 2002, 2003, 2011; Wada et al. 2004; Deringer et al. 2016; Ogawa et al. 2019; Sikorski et al. 2009; Naito et al. 2016) (Chen et al. 2014). Some authors regarded hydrogen bonds as the key factor governing the assembly of polymer chains, trying to explain many physical properties of cellulose. Recent studies have gradually reshaped this view. The impact of hydrogen bonds on the peeling-off of cellotetraose (Bergenstråhle et al. 2010) and the proposal of the hydrophobicity of cellulose (Lindman et al. 2010, 2021; Medronho et al. 2014; Glasser et al. 2012) both indicated that the contribution of hydrogen bonding interaction to the insolubility of cellulose was overemphasized.
After a few years of debate, it seems that we have established that other noncovalent interactions, such as electrostatic interaction and London dispersion interactions, are also responsible for the tight stacking of chains (Jarvis 2023). Recently, a detailed review evaluated the “exaggerated” role of hydrogen bonds (Wohlert et al. 2023) associated with the properties of cellulose in the past decades. Based on the linear tendencies of the heat of evaporation of analog molecules with numbers of hydroxy groups and molecular weights, the inter-chain hydrogen bond energy is 24 kJ/mol in crystalline cellulose Iβ (Nishiyama 2018), and London dispersion interaction is estimated to be 67 kJ/mol per glucose based on empirical Lennard-Jones potential parameters from the GLCYAM06 force field. Still, the first-principles-based quantification of the internal energy of cellulose and chitin is rare (Deringer et al. 2016). We previously quantified the partition of the noncovalent interaction in chitin and chitosan based on DFT-D2 calculations (Chen et al. 2021) using an energy decomposition analysis based on a low-dimension fragment approach (Deringer et al. 2016). Here, we have extended this method for the systematic analysis of cellulose, chitin, chitosan, and their allomorphs, as well as their monomers using three different generations of dispersion correction approaches (D2 (Grimme 2006), D3 (Grimme et al. 2010), D4 (Caldeweyher et al. 2017, 2019) in which polarizability was either considered or not. In addition, the upper limit of hydrogen bond strength is estimated based on the energy profile of hydroxy rotation, providing a basic, quantitative understanding of the noncovalent components of the crystalline polysaccharides.
Computational methods
Model construction
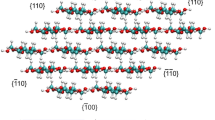
Eight types of crystalline polysaccharides whose atomic structures are available were considered, which were named cellulose Iα (Nishiyama et al. 2003), Iβ (Nishiyama et al. 2002), II (Langan et al. 1999, Chen et al. 2015), IIII(Wada et al. 2004), α-chitin-A (Sikorski et al. 2009; Deringer et al. 2016), α-chitin-B (Sikorski et al. 2009; Deringer et al. 2016), β-chitin (Nishiyama et al. 2011), chitosan (Naito et al. 2016). β-d-glucose and β-d-cellobiose (Jeffrey 1968). The unit cells and the corresponding fragments of these crystals are all presented in Fig. 1. The computational setup is identical to our former study (Chen et al. 2021), and a detailed description can be found in the following subsections.

Unit cell representation of cellulose, chitin, chitosan, glucose, and cellobiose. (All crystal structures will be deposited in glyco3D after publication)
Energy minimization
Periodic boundary conditions (PBC) based and dispersion-corrected DFT calculations were performed by using Quantum Espresso (QE) (Giannozzi et al. 2009, 2017) and VASP package (Kresse and Furthmüller 1996). D2 (Grimme 2006) and D3 (Grimme et al. 2010) are implemented in QE, and D4 (Caldeweyher et al. 2017, 2019) can be implemented in VASP (Hafner et al. 1997). The generalized gradient approximation (GGA) functional PBE (Perdew et al. 1996) was used for the geometry optimization of crystals. The total energy and force convergence thresholds for ion minimization were set to 1.0e-6 Ry and 1.0e-5 Ry/Bohr, respectively. The kinetic energy cutoff value of the wave function was 160 Ry. The k-points were set to (2, 2, 2, 0, 0, 0).
Energy decomposition analysis
The classical molecular mechanics (MM) represents the intermolecular noncovalent interaction as composed of Coulomb interactions between point charges and Lennard-Jones potential interactions of each paired atoms, including the London dispersion and Pauli repulsion terms. In DFT calculations, the intermolecular energy comprises terms such as electrostatics, exchange, induction, and dispersion interaction. For both cases, we can simplify them into dispersion energy and everything else as electrostatic interactions. The molecular interaction energy within a crystal can be written as Eq. (1).
Further decomposition for intra-chain and inter-chain terms results in four energy terms, which are the interchain electrostatic energy (Einter_E), the intrachain electrostatic energy (Eintra_E), the interchain dispersion energy (Einter_D), and the intrachain dispersion energy (Eintra_D) as presented in Eq. (2).
We rely on the low dimensional fragments approach widely used in DFT-based materials simulations (Deringer et al. 2016) to estimate the contribution of interchain interactions. In brief, DFT calculation was performed for the three-dimensional crystals (3D: raw unit cell applied with PBC) with relaxation for both atomic coordinates and crystal lattices, obtaining optimized energy and structure (noted as E3D_disp, corresponds to top left in Fig. 2). One isolated structural fragment (1D) was constructed by computationally “cleaving” the lattice apart from the above fully relaxed 3D structure. This is achieved by leaving one chain within the supercell and enlarging the transverse lattices by a factor of two, which “forcedly” separates this chain from its periodic boundary images, as shown in (top right of) Fig. 2. The total-energy computation was subsequently performed freezing both the box size and coordinates, and the obtained energy is noted as E1D_disp. This setup is slightly different from our previous work (Chen et al. 2022), in which the enlarged box and atoms were both relaxed, and makes the Eintra_E truly constant. In the previous studies, the Eintra_E was “assumed” to be unchanged, which was not the case, as seen in the comparison between Table 1 and Table S1. By switching off the dispersion correction and freezing the atoms both in the 3D system and in the 1D system, two other energy terms can be derived, noted as E3D_nodisp and E1D_nodisp, corresponding to the bottom left and right states in Fig. 2, respectively. This is illustrated in Eqs. (3), (4), and (5), respectively.
where N stands for the number of residues per unit cell.
The cohesion energy of crystals per residue equals the sum of Einter_D and Einter_E (Eq. (6)).
The graphical illustration of such energy decomposition for cellulose Iβ as a trial is shown in Fig. 2. On the graph, E1D_disp is the total energy of one isolated structural fragment with dispersion correction switched on. In contrast, E1D_nodisp is the one without dispersion correction. For E3D_disp and E3D_nodisp, the 3D subscript means a standard unit cell with PBC applied, and thus the crystal is three-dimensional and infinite. Disp. and Nodisp. indicated whether or not the dispersion correction is applied.

Illustration of the condensed 3D fragment and isolated 1D fragment with dispersion correction switched on/off used for the energy composition calculation

Comparison of intermolecular London dispersion (Einter_D) and electrostatic energies (Einter_E) (ECohe_E = Einter_D + Einter_E) based on DFT-D2 (left), D3(middle), and D4(right)
Results and discussion
Estimation of London dispersion interaction
Figure 3 and Table 1 show the decomposed energy for all crystals as a function of the generation of dispersion correction. The (Eintra_D, Einter_D, Einter_E, ECohe_E) of Iβ is estimated as (79, 62, 47, 109) kJ/mol per glucose based on DFT-D2, respectively. This quantification of Einter_D is close to the empirical estimation of 67 kJ/mol (Nishiyama 2018). Comparing the four allomorphs of cellulose within the D2 framework, the Einter_D of II is the largest (64 kJ/mol per residue), while that of IIII is the smallest (60 kJ/mol ), and Iα/Iβ is in the middle (62 kJ/mol).
Overall, the estimated Einter_D for eight polysaccharide crystals ranges from 60 to 74 kJ/mol, the Eintra_D varies from 76 to 105 kJ/mol, and the Einter_E varies from 47 to 70 kJ/mol per residue based on DFT-D2 calculation. The larger Einter_D of chitin than cellulose is due to the higher molecular weight of N-acetyl-glucosamine residue than glucose residue. If one normalizes the energy by the volume of the residue relative to cellulose Iβ, we obtained (62, 62, 64, 57, 51,55, 55, 51, 64) kJ/mol of EIntrer_D (Iα, Iβ, II, IIII, α-chitin-A, α-chitin-B, β-chitin, chitosan) (Table S2), showing smaller normalized EIntrer_D of chitin than cellulose. Both II and chitosan exhibited maximal values (64 kJ/mol). One can find (Table 1) in crystalline monomer and dimer that Eintra_D (59, 68 kJ/mol) and Einter_D (98, 79 kJ/mol) are, although different, already comparable to that of polymer crystals. The slightly higher value of Einter_D and smaller value of Eintra_D in small molecular crystal than polymer crystal is partially ascribed to the increased molecular weight of repeat units (162 for glucose residue, 171 for cellobiose and 180 for glucose) and partially because of the chain polarity, which can be either parallel or antiparallel.
Different number of hydroxy groups per residue also lead to different numbers of inter-molecular hydrogen bonds per residue, which are 5 for glucose, 4 for cellobiose, 2 for II, IIII, chitin, and 1 for Iα/Iβ and chitosan. Because the nature of hydrogen bond is mostly electrostatic interaction, the Einter_E in small molecular crystals is much larger than its Einter_D (Fig. S1) and Einter_E of polymer crystals, respectively. We simply divide Einter_E by the number of hydrogen bonds per residue for a quick and rough calculation. The strength of the single hydrogen bond in mono and dimer crystals can be estimated to be under 30 kJ/mol in glucose and 27 kJ/mol in cellobiose, respectively. Similarly, the Einter_E in cellulose II, IIII, and chitin are larger than Iα, Iβ, and chitosan, due to one more inter-chain hydrogen bond per residue. More measurements of hydrogen bond interactions will be discussed in the last section.
Based on the DFT-D2 calculation, for the β-1,4-linked crystalline polysaccharides, the London dispersion interaction represents by 48 ~ 58% of the total cohesion energy of the polymer crystal regardless of polymer categories and types of correction used, as can be overviewed in Fig. 3 (left), Table 2, S4, and S5.
The impact of three generations of dispersion correction
When dispersion correction was modulated from D2 to the other two generations (D3 and D4), the four energy terms (Eintra_D, Einter_D, Einter_E, ECohe_E) of cellulose Iβ varied to (52, 53, 51, 105 kJ/mol) for D3 and (79, 49, 52, 100 kJ/mol) for D4, showing the monotonical decreasing of Einter_D and the increase of Einter_E and resulting in the Einter_E slightly over Einter_D. Such a reverse trend between Einter_D and Einter_E is also applicable to other crystals, as presented in Fig. S2. In D2, the dispersion coefficients for each atom species were constant, no matter their chemical contexts and the atomic number of atoms. In D3 and D4, the local electron polarizability effect was accounted for, and the dispersion coefficients were automatically adjusted according to their local chemical environment. In detail, the atomic partial charge used for scaling of polarizabilities relies on Mulliken partial charge in D3, but relies on the electronegativity equilibration partial charge in D4. Such treatment results in more expensive calculations and different energy values. By comparing with the benchmark of available molecular dipole-dipole dispersion coefficients, the D4 achieved a slightly better agreement with the experiment (Caldeweyher et al. 2019).
The update of dispersion correction leads to a slight difference in predicted unit cell parameters and thus also minor differences in chain packing (Table S3), especially the slight expansion of unit cell parameters a and b, which reflected the relatively less tight packing of pyranose ring in D3 and D4, and therefore reduced Einter_D and increased Einter_E. Still, the Einter_D takes 41 ~ 55% of the total intermolecular interactions for D3, and 35 ~ 48% for D4, as shown in Table S4.
No matter which type of dispersion correction is used, the Einter_D and Eintra_D of chitin are always higher than those of cellulose. This is simply ascribed to the larger molecular weight of the repeat unit (162 Da for cellulose and 203 Da for chitin). Nominalization by volume will reduce the Einter_D and Eintra_D of chitin, as previously discussed.
Estimations of hydrogen bond strength
Although more than one hundred years have passed since the first proposal for hydrogen bonds (Huggins1971; Derewenda et al. 2021), the estimation of its range of strength is still under development (Emamian et al. 2019). To computationally estimate one hydrogen bond interaction between small molecules such as water, one can separate the hydrogen bonding paired molecules and estimate the energy difference as hydrogen bond strength since the London dispersion interaction between paired small molecules is small and thus can be neglected. However, one cannot simply do so for hydrogen bonds in cellulose or chitin because of other electrostatic interactions and the increased London dispersion due to increased molecular weight. In addition, dividing Einter_E by the number of interchain hydrogen bonds would overestimate the hydrogen bond contribution because other multipolar electrostatic interactions also contribute to Einter_E. In the textbooks (Mark 2023), the hydrogen bonds are often judged by an arbitrary geometric factor: donor (H)-acceptor length < 0.27 nm and H-donor-acceptor angle < 30º. When such criteria are not fulfilled, they fall into the category of Coulomb interaction. Based on this, we developed an approach by extracting one chain or sheet out of the 3D crystal (as shown in Fig. 4) and rotating the hydroxy group around the C-O bond. Single-point energy calculations were run at each point by freezing all the atoms. Only a proton is moving, so the London dispersion interaction can be regarded as nearly constant, and the energy difference can be regarded as an indicator of hydrogen bond and partial contribution from electrostatic repulsion interaction. This method is similar to the study by Estácio et al. (Estácio et al. 2004), which shows that this hydrogen bond energy value is overestimated and can be considered an upper limit of hydrogen bond strength. Details for each hydrogen bond energy in β-chitin are also provided in Fig. S3.
Taking the intra-chain hydrogen bond of Iβ as an example, the H-O3 was rotated around the O3-C3 bond with a stepwise increment of 10º starting from the initial energy minimum (labeled as A), and single-point energy was calculated at each frame, as shown in Fig. 4. The variation of total energy is purely ascribed to the movement of hydrogen atom. The difference between the optimized energy and when H-O…O angle becomes 30º is considered as the strength of intra-chain O3-H…O5 hydrogen bond (labeled B in Fig. 4a). The estimation of another intra-chain (O2-H…HO6) hydrogen bond was done in a similar way by applying rotation for O2-H around the C2-O2 bond as shown in Fig. 4b. The estimation of inter-chain hydrogen bond (O6-H…O3) requires the simultaneous rotations of C2-O2-H and C6-O6-H angles because the sole rotation of C6-O6-H will induce unreasonable short HO6…HO2 contact (< 1 Å). To avoid this short proton-proton contact, the HO2 hydroxy and HO6 hydroxy groups are rotated in the opposite direction in the red arrow in Fig. 4c. The rotation of HO2 in Fig. 4c and b follow the same direction. The total energy variation of the simultaneous rotation of HO2 and HO6 was subtracted by the energy profiles of the sole HO2 rotation in Fig. 4b, and the result is shown in Fig. 4c. The estimation of other crystals follows the receipt of Iβ.

Total energy variation of Iβ chain as the function of H-O rotated angle around bond C-O. The arrow indicates the rotation trajectory of hydroxy groups. A, B, C, D, E in energy profiles corresponds to the same label in molecular snapshots, which indicates the selected frames during the rotation trajectory of hydroxy groups. The red arrows indicate the rotational direction of hydroxy groups from 0 to 360 degrees
By setting the universal hydrogen bond criteria (H…acceptor < 0.27 nm and H-D-A < 30º), the three major hydrogen bonds (EHO3…O5, EHO2…O6, EHO6…O23) of Iβ can be qualitatively estimated to have the upper limits of 25 kJ/mol, 24 kJ/mol, and 31 kJ/mol, with DFT-D2 as labeled in the dashed line in Fig. 4a and b, and 4c. The slightly higher value of EHO6…O3 than the other two can be ascribed to the additional contribution of electrostatic attraction from the O6H…O2 pair since the rupture of O6H…O3 also alters the O6H…O2 distance during the rotation of the hydroxy group (Fig. 4 & S4). Summary of all hydrogen bond strength is provided in Table 3, showing the range from 14 to 33 kJ/mol, which is similar to that in the estimation of alcohol hydrogen bond strength (24 kJ/mol) of small analogs (Nishiyama 2018). This indicates that the hydrogen bond strength in cellulose, chitin, and chitosan is not particularly strong but is similar to their smaller analogs (such as glucose, Table S6). For chitin and chitosan, the strength of a single NH…OC hydrogen bond is 26 and 34 kJ/mol, respectively, within the same magnitude of the strength of OH…OH hydrogen bond of cellulose. All the hydrogen bond strength estimation depends very little on the generation types of London dispersion correction, as shown in Table S7, S8 (13 ~ 34 kJ/mol for D3), and S9 (14 ~ 32 kJ/mol for D4) for all crystals. These precise estimations of OH…OH strength is also identical to the rough estimations of glucose and cellobiose in the previous section.
Each glucose residue in cellulose Iβ contains three free hydroxy groups that form one inter-chain hydrogen bond and two intra-chain hydrogen bonds on average, the strength of which is < 31 and < 50 kJ/mol per glucose, proving that both the inter- and intra-chain hydrogen bonds are weaker than the corresponding inter- and intra-chain London dispersion interactions (62 and 79 kJ/mol per glucose), respectively (Fig. 5). This picture is regardless of types of dispersion correction (52, 51, 53, 27 of Eintra_D, Eintra_HB, Einter_D, Einter_HB for D3 and 79, 53, 49, 31 for D4). A similar expression also applies to cellulose Iα, exhibiting similar structural features as Iβ, as shown in Table 3.

Comparison of (inter-chain and intra-chain) London dispersion interaction and hydrogen bond strength in crystals
For other cellulose allomorphs (II and IIII) and chitin, as well as chitosan, the intra-chain hydrogen bond (O3-H...O5O3) is retained. However, the planar hydrogen-bond network disappears due to the conformational variation of the exocyclic hydroxymethyl group, which is tg in native cellulose, gg/gt in α-chitin, and gt in the rest. The number of inter-chain hydrogen bonds increases from 1 to 2 per residue, and the intra-chain one decreases from 2 to 1, thus accompanied by the increased total strength of inter-chain hydrogen bonds. However, the strength of inter-chain hydrogen bonds is always below 50 kJ/mol from D2 and D3 and 47 kJ/mol from D4, showing that none exceeds their corresponding inter-chain London dispersion interaction (see Table 2). Regarding the intra-chain interactions, hydrogen bonds contribute far less than dispersion interaction (< 32 kJ/mol versus > 50 kJ/mol, respectively), as also shown in Table 2. The same picture between intra- and inter-chain dispersion interactions and hydrogen bonds also holds for other polysaccharide analogs. As shown in Table S10, the relative ratio of hydrogen bonding interaction occupied in the total interchain interaction in crystals varies between 23% and 40%. In comparison, London dispersion energy fluctuates between 35% and 58% (Table S4), and the overall electrostatic interactions occupy from 42 to 61% (Table S5).
When hydroxy groups are rotated around their corresponding C-O bond (Fig. 4), the energy barrier during the C-O-H angle rotation may reach 50 ~ 60 kJ/mol per residue. One may simply think this should be considered as the hydrogen bond strength. Such a thought is improper because the energy variation induced by the rotation of the hydroxy group includes both hydrogen bonds and other repulsions or attractions between hydrogen and nearby atoms. This repulsion or attraction occurs in polymers but not for small molecules (such as water) in an isolated state due to the steric effect of adjacent atoms in the polymer chain. One obvious evidence is the bimodal shape of the energy profile, where an energy minimum occurs between 200º and 250º, and is due to the electrostatic attraction between mobile hydrogen and its adjacent oxygen. Their geometry parameters, at this minimum, are far beyond the standard hydrogen bond criteria. At this low energy minimum, the nearby electrostatic repulsion is the smallest. The energy variation is consistently lower than 32 kJ/mol in comparison to the initial state (20 kJ/mol in Fig. 4a and 32 kJ/mol in Fig. 4b and 18 kJ/mol in Fig. 4c), which is much smaller than the energy barrier. Therefore, our estimated energy difference is already the upper limit of hydrogen bond strength.
The hydrogen bond strength estimated here is slightly higher than those reported for small molecules. This is because one hydroxy in polysaccharide crystal structures acts as both donor and acceptor due to the hydrogen bonding network, constructing correlation among each hydrogen bond. Influencing one may also partially interrupt others. The cooperativity in the hydrogen bond network enhances the strength of a single hydrogen bond, but the extent is limited (Qian 2008; Masella et al. 2000). The hydrogen bond strength is also context-dependent in protein, which is stronger in the inner hydrophobic core than the surface, but never found to dominate the structural stability (Deechongkit et al. 2004). According to our estimation on hydrogen bond and London dispersion interaction, a similar principle can be applied to crystalline polysaccharides, .
Conclusion
To summarize, using the DFT calculation and modulation of dispersion correction and the single energy calculation with rotating hydroxy groups, we have systematically quantified the London dispersion interaction and strength of hydrogen bonds of cellulose, chitin, and chitosan and their monomers and dimers. We can confirm that inter-chain London dispersion interaction exceeds the strength of inter-chain hydrogen bonds within the lattice energy of cellulose Iβ as Nishiyama reported. In addition, the intra-chain London dispersion interaction was also proved to be stronger than the intra-chain hydrogen bonds for Iβ crystals. Moreover, these findings not only apply to cellulose Iβ but also can be extended to other cellulose allomorphs (Iα, II, IIII) and other β-(1,4)-crystalline polysaccharides (chitin and chitosan). The alteration of different generations of dispersion interactions slightly alters the absolute value of intermolecular dispersion and electrostatic and hydrogen bonding energies due to slightly different chain packing and unit cell parameters compared to experimental observation. Still the picture that London dispersion interaction exceeds hydrogen bonding interaction always stands, for both inter- and intra-chain terms. Overall, the inter-chain hydrogen bonding interaction occupied 23 ~ 40% of the total interchain molecular interaction. At the same time, London dispersion energy fluctuated between 35% and 58%, and the electrostatic interactions occupied from 42 to 61% among eight crystals.
Our finding offers molecular insights to understand the driving force for the initial assembly of polymer chains during the biosynthesis of cellulose nanofibrils. Our quantification also provides direct evidence that refutes the hydrogen bonding interaction dominated dissolution mechanism of cellulose and chitin. One may argue that although one hydrogen bond is not strong enough, the activation energy to peel off abundant hydrogen bonds along a polymer chain would be strong enough. However, the peeling-off energy required against London dispersion interaction in the crystal would be also higher than the total energy of the many hydrogen bonds. In the future, our approach can be extended to the co-crystal between cellulose/chitin/chitosan and small molecules, such as cellulose/ammonia and cellulose/EDA complex, understanding the energy components and providing insight to develop a protocol for the deconstruction of these crystals.
Data availability
Not applicable.
Change history
05 March 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10570-024-05752-9
20 May 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10570-024-05937-2
References
Bergenstråhle M, Wohlert J, Himmel ME, Brady JW (2010) Simulation studies of the insolubility of cellulose. Carbohydr Res 345(14):2060–2066. https://doi.org/10.1016/j.carres.2010.06.017
Caldeweyher E, Bannwarth C, Grimme S (2017) Extension of the D3 dispersion coefficient model. J Chem Phys 147(3):034112. https://doi.org/10.1063/1.4993215
Caldeweyher E, Ehlert S, Hansen A, Neugebauer H, Spicher S, Bannwarth C, Grimme S (2019) A generally applicable atomic-charge dependent London dispersion correction. J Chem Phys 150(15):154122. https://doi.org/10.1063/1.5090222
Chen P, Nishiyama Y, Putaux J-L, Mazeau K (2014) Diversity of potential hydrogen bonds in cellulose I revealed by molecular dynamics simulation. Cellulose 21(2):897–908. https://doi.org/10.1007/s10570-013-0053-x
Chen P, Nishiyama Y, Wohlert J (2021) Quantifying the influence of dispersion interactions on the elastic properties of crystalline cellulose. Cellulose 28(17):10777–10786. https://doi.org/10.1007/s10570-021-04210-0
Chen P, Ogawa Y, Nishiyama Y, Bergenstråhle-Wohlert M, Mazeau K (2015) Alternative hydrogen bond models of cellulose II and IIII based on molecular force-fields and density functional theory. Cellulose 22(3):1485–1493. https://doi.org/10.1007/s10570-015-0589-z
Chen P, Zhao CJ, Wang HY et al (2022) Quantifying the contribution of the dispersion interaction and hydrogen bonding to the anisotropic elastic properties of Chitin and Chitosan. Biomacromolecules 23(4):1633–1642. https://doi.org/10.1021/acs.biomac.1c01488
Deechongkit S, Nguyen H, Powers E et al (2004) Context-dependent contributions of backbone hydrogen bonding to β-sheet folding energetics. Nature 430:101–105. https://doi.org/10.1038/nature02611
Derewenda ZS (2021) On the centennials of the discoveries of the hydrogen bond and the structure of the water molecule: the short life and work of Eustace Jean Cuy (1897–1925). Acta Crystallogr Sect A. https://doi.org/10.1107/S205327332100
Deringer VL, Englert U, Dronskowski R (2016) Nature, strength, and cooperativity of the hydrogen-bonding network in α-chitin. Biomacromolecules 17(3):996–1003. https://doi.org/10.1021/acs.biomac.5b01653
Emamian S, Lu T, Kruse H, Emamian H (2019) Exploring Nature and Predicting Strength of Hydrogen Bonds: a correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J Comput Chem 40:2868–2881. https://doi.org/10.1002/jcc.26068
Estácio SG, Cabral do Couto P, Costa Cabral BJ et al (2004) Energetics of Intramolecular Hydrogen Bonding in Di-substituted Benzenes by the ortho-para Method. J Phys Chem A 108(49):10834–10843. https://doi.org/10.1021/jp0473422
Giannozzi P, Andreussi O, Brumme T et al (2017) Advanced capabilities for materials modelling with Quantum ESPRESSO. J Phys-Condens Mat 29(46):465901. https://doi.org/10.1088/1361-648X/aa8f79
Giannozzi P, Baroni S, Bonini N et al (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys Condens Matter 21(39):395502. https://doi.org/10.1088/0953-8984/21/39/395502
Glasser WG, Atalla RH, Blackwell J, Malcolm BR et al (2012) About the structure of cellulose: debating the Lindman hypothesis. Cellulose 19(3):589–598. https://doi.org/10.1007/s10570-012-9691-7
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 27(15):1787–1799. https://doi.org/10.1002/jcc.20495
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104. https://doi.org/10.1063/1.3382344
Hafner J, Kresse G (1997) The Vienna AB-Initio Simulation Program VASP: an efficient and versatile Tool for studying the Structural, dynamic, and Electronic Properties of materials. In: A Gonis, A Meike, P.E.A. Turchi (Eds) Prop Complex Inorg Solids pp. 69–82. Boston: Springer. https://doi.org/10.1007/978-1-4615-5943-6_10
Huggins ML (1971) 50 years of hydrogen bond theory. Angew Chem Int Ed Engl 10(3):147–152. https://doi.org/10.1002/anie.197101471
Jarvis MC (2023) Hydrogen bonding and other non-covalent interactions at the surfaces of cellulose microfibrils. Cellulose 30:667–687. https://doi.org/10.1007/s10570-022-04954-3
Jeffrey G (1968) The refinement of the crystal structures of β--glucose and cellobiose. Acta Crystallogr., Sect. B: Structural Crystallography and Crystal Chemistry 24(6):830–838. https://doi.org/10.1107/S0567740868003250
Kresse G, Furthmüller J (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp Mater Sci 6(1):15–50. https://doi.org/10.1016/0927-0256(96)00008-0
Langan P, Nishiyama Y, Chanzy H (1999) A revised structure and hydrogen-bonding system in cellulose II from a neutron fiber diffraction analysis. J Am Chem Soc 121(43):9940–9946. https://doi.org/10.1021/ja9916254
Lindman B, Karlström G, Stigsson L (2010) On the mechanism of dissolution of cellulose. J Mol Liq 156(1):76–81. https://doi.org/10.1016/J.MOLLIQ.2010.04.016
Lindman B, Medronho B, Alves L, Norgren M, Nordenskiöld L (2021) Hydrophobic interactions control the self-assembly of DNA and cellulose. Q Rev Biophys 54:33541444. https://doi.org/10.1017/S0033583521000019
Mark A, Andrey A, Cathrine B et al (2023) GROMACS 2023 manual (version 2023). Zenodo. https://doi.org/10.5281/zenodo.7588711
Masella M, Flament JP (2000) Influence of cooperativity on hydrogen bond networks. Mol Simul 24(1–3):131–156. https://doi.org/10.1080/08927020008024192
Medronho B, Lindman B (2014) Competing forces during cellulose dissolution: from solvents to mechanisms. Curr Opin Colloid Interface Sci 19(1):32–40. https://doi.org/10.1016/j.cocis.2013.12.001
Naito PK, Ogawa Y, Sawada D, Nishiyama Y, Iwata T, Wada M (2016) X-ray crystal structure of anhydrous chitosan at atomic resolution. Biopolymers 105(7):361–368. https://doi.org/10.1002/bip.22818
Nishiyama Y (2018) Molecular interactions in nanocellulose assembly. Philos Trans Royal Soc A 376(2112):20170047. https://doi.org/10.1098/rsta.2017.0047
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124(31):9074–9082. https://doi.org/10.1021/ja0257319
Nishiyama Y, Noishiki Y, Wada M (2011) X-ray structure of anhydrous β-chitin at 1 Å resolution. Macromolecules 44(4):950–957. https://doi.org/10.1021/ma102240r
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose Iα from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125(47):14300–14306. https://doi.org/10.1021/ja0257319
Ogawa Y, Naito P-K, Nishiyama Y (2019) Hydrogen-bonding network in anhydrous chitosan from neutron crystallography and periodic density functional theory calculations. Carbohydr Polym 207:211–217. https://doi.org/10.1016/j.carbpol.2018.11.042
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77(18):3865–3868. https://doi.org/10.1103/physrevlett.77.3865
Qian X (2008) The effect of cooperativity on hydrogen bonding interactions in native cellulose Iβ from ab initio molecular dynamics simulations. Mol Simul 34(2):183–191. https://doi.org/10.1080/08927020801961476
Sikorski P, Hori R, Wada M (2009) Revisit of alpha-chitin crystal structure using high resolution X-ray diffraction data. Biomacromolecules 10(5):1100–1105. https://doi.org/10.1021/bm801251e
Wada M, Chanzy H, Nishiyama Y (2004) Cellulose III_I crystal structure and hydrogen bonding by synchrotron X-ray and neutron fiber diffraction. Macromolecules 37(23):8548–8555. https://doi.org/10.1021/ma0485585
Wohlert M, Benselfelt T, Wågberg L, Furó I, Berglund LA, Wohlert J (2023) Cellulose and the role of hydrogen bonds: not in charge of everything. Cellulose 29(1):1–23. https://doi.org/10.1007/s10570-021-04325-4
Acknowledgments
PC thanks the Natural Science Foundation of China (52373097) and Beijing Natural Science Foundation (2232064). This work was partially supported by State Key Laboratory of Pulp and Paper Engineering (202306).
Funding
This research was supported by the Natural Science Foundation of China (52373097) and Beijing Natural Science Foundation (2232064). This work was partially supported by State Key Laboratory of Pulp and Paper Engineering (202306).
Author information
Authors and Affiliations
Contributions
YL & CY: Investigation, data analysis, original draft. Y C & XH: Investigation. ZS & XL: Resources and review. HQ & Y. Nishiyama & TH: Supervision and review. PC: Project administrator, investigation, supervision, original draft and review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
All authors state that they adhere to the Ethical Responsibilities of Authors. In addition, this article does not contain any studies with human participants or animal performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Acknowledgments and Funding section are updated.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Yan, C., Chen, Y. et al. The major role of London dispersion interaction in the assembly of cellulose, chitin, and chitosan. Cellulose 30, 8127–8138 (2023). https://doi.org/10.1007/s10570-023-05376-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-023-05376-5