
Abstract
This paper contains the synthesis of the new 2-morpholinoethyl substituted bis-(NHC)Pd(II) complexes and their catalytic activity in direct arylation reaction. The new bis-(NHC)Pd(II) complexes have been prepared from Ag(I)NHC complexes by using transmetallation method. The new bis-(NHC)Pd(II) complexes have been characterized by using 1H NMR, 13C NMR, FTIR spectroscopy and elemental analysis techniques. Molecular and crystal structure of the complex 1a and its ligand (N-benzylbenzimidazole) were obtained by single crystal X-ray diffraction method. The new bis-(NHC)Pd(II) complexes exhibit activity in the reaction after being examined as catalysts in the direct arylation (C–H activation) reaction.
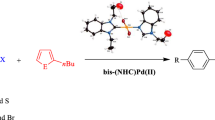
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The N-heterocyclic carbenes (NHCs) were known as unstable and non-isolable intermediates when first discovered by Wanzlick [1] and Öfele [2] in 1968. However, after the report on the extraordinary stability, isolation, and storability of crystalline NHC IAd by Arduengo et al. in 1991 [3,4,5], numerous studies have been reported on the NHC as ligands in organic and organometallic chemistry [6]. These ligands with strong σ-donor and weak π-acceptor properties can form stable complexes with almost all transition metals [7,8,9].
In recent years NHC ligands have an important place in transition-metal chemistry due to their unique complexation properties, synthetic versatility and highly adjustable properties [10]. Metal-NHC complexes, especially Ag(I)-NHC complexes have attracted continuous attention [11]. They are used as suitable carbene transfer reagents for other metal–carbene complexes (as Pd, Ru, Ni, Rh, and Ir) which are not always easily found [12,13,14,15,16]. Also, biomedical applications of Ag-NHC complexes such as antimicrobial, anticancer and antitumor agents have been researched [17,18,19].
Pd-NHC complexes are considered to be one of the most important classes of metal-NHC complexes. The pioneering participation from the groups of Nolan [20], Herrmann [21, 22], Glorious [23] and Organ [24] have been mentioned in this topic. Metal-NHC complexes have found wide application area that including olefin metathesis, hydrosilylation, and Pd-catalyzed cross-coupling reactions [25]. Bis-(NHC)Pd(II)dihalide complexes are efficient catalysts for different C–C and C–N coupling reactions [26, 27]. Usually accepted that the chelate effect revealed on their metal complexes presents extra stability for the generation of durable metal complexes, which are extremely desirable for catalytic applications, especially those requiring harsh reaction conditions [28].
The palladium catalyzed direct arylation of isoxazoles [29], the reaction of the five-membered heteroaromatic ring with aryl halides via C–H bond activation, has been firstly reported by Nakamura et al in 1982 [30,31,32,33,34,35,36,37,38,39]. One of the most important methods in modern organic chemistry is palladium-catalyzed carbon–carbon (C–C) cross-coupling reactions since direct (C–H) arylation has taken attention as an alternative C–C coupling reaction in recent years [37, 40]. Recently, reactions of the C–H activation via organopalladium intermediate species have become multipurpose [41, 42]. Palladium catalytic systems are rather widespread owing to their selectivity, high activity, versatility, and efficiency. Also, different catalytic systems for the C–H bond transformations have been improved for the eco-friendly synthesis method [43,44,45].
In recent years, numerous studies have been published on the metal-NHC complexes containing direct arylation, transfer hydrogenation, Suzuki–Miyaura and Heck reactions [46,47,48,49,50,51,52,53,54]. Our study contains the synthesis of the new series of 2-morpholinoethyl-substituted bis-(NHC)Pd(II) complexes and their structural and spectroscopic characterization. The structure of 1a was confirmed by the single-crystal X-ray diffraction method. Also, the catalytic activities of the bis-(NHC)Pd(II) complexes have been investigated that they are more efficient and stable catalysts for the direct arylation reactions of 2-n-butylfuran and 2-n-butylthiophene with aryl bromide.
2 Experimental
All synthesis involving bis-(NHC)Pd(II) complexes 1a–f were carried out under an inert atmosphere in flame-dried glassware using standard Schlenk techniques. The solvents used were purified by distillation over the drying agents indicated and were transferred under Ar: Et2O (Na/K alloy), CH2Cl2 (P4O10), hexane, toluene (Na).
All other reagents were commercially available from Aldrich Chemical Co. and used without further purification. Melting points were identified in glass capillaries under air with an Electrothermal-9200 melting point apparatus. FT-IR spectra were saved in the range 400–4000 cm−1 on Perkin Elmer Spectrum 100 FT-IR spectrometer. Proton (1H) and Carbon (13C) NMR spectra were recorded using either a Varian AS 300 Merkur spectrometer operating at 300 MHz (1H), 75.47 MHz (13C) in CDCl3 with tetramethylsilane as an internal reference. All reactions were observed on an Agilent 6890 N GC system by GC-FID with an HP-5 column of 30 m length, 0,32 mm diameter and 0,25 μm film thickness. Column chromatography was performed using silica gel 60 (70–230 mesh). Elemental analyses were performed by İnönü University Scientific and Technological Research Center (Malatya, Turkey).
Single crystal X-ray diffraction data set of the complex 1a and its ligand (N-benzylbenzimidazole) were collected at room temperature on a Rigaku-Oxford Xcalibur EOS diffractometer using graphite monochromated Mo–Kα radiation (λ = 0.71073 Å). The data were collected and integrated using CrysAlisPro software [55]. Utilizing OLEX2 [56], the structures were solved by direct methods in SHELXT [57] and refined by full-matrix least-squares on F 2 in SHELXL [58]. Anisotropic thermal parameters were applied to all non-hydrogen atoms. Hydrogen atoms of the ligand were found in the difference map and refined freely. For the complex, all hydrogen atoms were placed in geometrically idealized positions (C–H = 0.93–0.97 Å, Cl–H = 1.630–1.707 Å). Crystallographic data is summarized in Table 1.
2.1 Synthesis of bis[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene]dichloropalladium(II), 1a
PdCl2(PhCN)2 (0.078 g, 0.3 mmol) was added to a solution of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I) (288 mg, 0.6 mmol) in dichloromethane (20 mL). The reaction mixture was stirred for 24 h at room temperature in the dark conditions. Then filtered through Celite and the solvents were evaporated under vacuum to afford the product as a light yellow solid. The crude product was recrystallized from dichloromethane/diethyl ether (1:3) at room temperature. Yield: 0.172 g (70%). m.p.: 205–206 °C; ν(CN): 1452.6 cm−1. Anal. Calc. for C40H46N6O2PdCl2: C: 58.58; H: 5.65; N: 10.25. Found: C:59.41; H: 6.13; N: 9.82. 1H NMR (300 MHz, CDCI3, δ, ppm); 2.42 (t, 8H, J: 4.2 Hz, –NCH 2 CH2O–); 3.11 (m, 4H, –NCH2CH 2 NC4H8O); 3.48 (m, 8H, –NCH2CH 2 O–); 5.00 (t, 4H, J: 4.5 Hz, –NCH 2 CH2NC4H8O–); 6.18 (s, 4H, –CH 2 C6H5–); 7.03–7.35 (m, 18H, Ar–H). 13C NMR (75 MHz, CDCI3, δ, ppm); 52.1 (–NCH2CH2O–); 54.3 (–NCH2 CH2NC4H8O); 58.0 (–NCH2 CH2O–); 66.9 (–NCH2CH2NC4H8O–); 67.3 (–C6H4 CH2–); 111.0, 111.7, 111.9, 124.2, 128.1, 133.0, 133.2, 134.1, 134.2, 134.5, and 137.6 (Ar–C); 181.6 (2-CH).
2.2 Synthesis of Bis[1-(3-methylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] dichloropalladium(II), 1b
The synthesis of 1b was carried out in the same way as that described for 1a, but chloro[1-(3-methylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I) (296 mg, 0.6 mmol) was used instead of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I). Yield: 0.188 g, (74%). m.p.: 254–256 °C; ν(CN): 1456.7 cm−1. Anal. Calc. for C42H50N6O2PdCl2: C: 59.47; H: 5.94; N: 9.91. Found: C: 59.61; H: 6.01; N: 9.93. 1H NMR (300 MHz, CDCI3, δ, ppm); 2.30 (s, 6H, –C6H4–CH 3 ); 2.99 (t, 8H, J: 4.5 Hz, –NCH 2 CH2O–); 3.36 (t, 4H, J: 7.6 Hz, –NCH2CH 2 NC4H8O); 3.64 (t, 8H, J: 4.5 Hz, –NCH2CH 2 O–); 4.89 (t, 4H, J: 7.6 Hz, –NCH 2 CH2NC4H8O–); 6.14 (s, 4H, –CH 2 C6H4–); 7.03–7.39 (m, 16H, Ar–H). 13C NMR (75 MHz, CDCI3, δ, ppm); 45.7 (–C6H4–CH3); 52.8 (–NCH2CH2O–); 53.8 (–NCH2 CH2NC4H8O); 57.7 (–NCH2 CH2O–); 66.8 (–NCH2CH2NC4H8O–); 67.1 (–C6H4 CH2–); 110.4, 110.7, 111.5, 123.0, 127.8 128.6 and 129.3 (Ar–C); 181.8 (2-CH).
2.3 Synthesis of Bis[1-(4-methylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] dichloropalladium(II), 1c
The synthesis of 1c was carried out in the same way as that described for 1a, but chloro[1-(4-methylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I) (296 mg, 0.6 mmol) was used instead of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I). Yield: 0.198 g, (78%). m.p.: 249–251 °C; ν(CN): 1448.1 cm−1. Anal. Calc. for C42H50N6O2PdCl2: C: 59.47; H: 5.94; N: 9.91. Found: C: 59.53; H: 5.88; N: 9.82. 1H NMR (300 MHz, CDCI3, δ, ppm); 2.36 (s, 6H, –C6H4–CH 3 ); 2.42 (t, 8H, J: 4.2 Hz, –NCH 2 CH2O–); 3.11 (m, 4H, –NCH2CH 2 NC4H8O); 3.47 (m, 8H, –NCH2CH 2 O–); 4.99 (t, 4H, J: 4.5 Hz, –NCH 2 CH2NC4H8O–); 6.20 (s, 4H, –CH 2 C6H4–); 7.02–7.18 (m, 16H, Ar–H). 13C NMR (75 MHz, CDCI3, δ, ppm); 21.2 (–C6H4–CH3); 52.1 (–NCH2CH2O–); 54.2 (–NCH2 CH2NC4H8O); 57.6 (–NCH2 CH2O–); 66.8 (–NCH2CH2NC4H8O–); 67.4 (–C6H4 CH2–); 110.4, 111.4, 111.6, 123.1, 127.7 and 129.3 (Ar–C); 181.8 (2-CH).
2.4 Synthesis of Bis[1-(2,3,4,5,6-pentamethylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] dichloropalladium(II), 1d
The synthesis of 1d was carried out in the same way as that described for 1a, but chloro[1-(2,3,4,5,6-pentamethylbenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I) (330 mg, 0.6 mmol) was used instead of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I). Yield: 0.207 g, (72%). m.p.: 248–250 °C; ν(CN): 1456.7 cm−1. Anal. Calc. for C50H66N6O2PdCl2: C: 62.53; H: 6.93; N: 8.75. Found: C: 62.64; H: 7.02; N: 8.67. 1H NMR (300 MHz, CDCI3, δ, ppm); 2.30 (s, 6H, –C6–CH 3 ); 2.38–2.40 (s, 30H, –C6–(CH 3 )5); 2.42 (s 8H, –NCH 2 CH2O–); 2.60 (m, 4H, –NCH2CH 2 NC4H8O); 3.50 (s, 8H, –NCH2CH 2 O–); 3.78 (m 4H, –NCH 2 CH2NC4H8O–); 5.12 (s, 4H, –CH 2 C6(CH3)5–); 7.35–7.47 (m, 8H, Ar–H). 13C NMR (75 MHz, CDCI3, δ, ppm); 16.9–17.6 (–C6–(CH 3 )4); 17.3 (–C6–CH 3 ); 51.3 (–NCH 2 CH2O–); 51.7 (–NCH2CH 2 NC4H8O); 54.0 (–NCH2CH 2 O–); 65.8 (–NCH 2 CH2NC4H8O–); 66.8 (–C6(CH3)5CH 2 –). 127.9, 133.1, 134.3, 134.5, 134.9 and 135.9 (Ar–C); 182.0 (2-CH).
2.5 Synthesis of Bis[1-(3,4,5-trimethoxybenzyl)-3-(2-morpholinoethyl)benzimidazol-2-ylidene] dichloropalladium(II), 1e
The synthesis of 1e was carried out in the same way as that described for 1a, but chloro[1-(3,4,5-trimethoxybenzyl)-3-(2-morpholinoethyl)-2-ylidene]silver(I) (342 mg, 0.6 mmol) was used instead of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I). Yield: 0.210 g, (70%). m.p.: 225–238 °C; ν(CN): 1455.2 cm−1. Anal. Calc. for C46H58N6O8PdCl2: C: 55.23; H: 5.84; N: 8.40. Found: C: 55.38; H: 5.72; N: 8.21. 1H NMR (300 MHz, CDCI3, δ, ppm); 2.42 (t, 8H, J: 4.2 Hz, –NCH 2 CH2O–); 3.12 (m, 4H, –NCH2CH 2 NC4H8O); 3.51 (s, 8H, –NCH2CH 2 O–); 3.86 (s, 6H, –C6H2OCH 3 ); 3.89 (s, 12H, –C6H2(OCH 3 )2); 5.01 (m 4H, –NCH 2 CH2NC4H8O–); 6.13 (s, 4H, –CH 2 C6H2(OCH3)3–). 13C NMR (75 MHz, CDCI3, δ, ppm); 52.3 (–NCH2CH2O–); 54.2 (–NCH2 CH2NC4H8O); 56.4 (–C6H2OCH3); 56.7 (–C6H2(OCH3)2); 57.8 (–NCH2 CH2O–); 60.8 (–NCH2CH2NC4H8O–); 66.8 (–CH2C6H2(OCH3)3–); 131.1, 133.3, 133.9, 134.9, 135.0,153.4 and 153.8 (Ar–C); 181.7 (2-CH).
2.6 Synthesis of Bis[1-butyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene]dichloropalladium(II), 1f
The synthesis of 1f was carried out in the same way as that described for 1a, but chloro[1-butyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I) (267 mg, 1.2 mmol) was used instead of chloro[1-benzyl-3-(2-morpholinoethyl)benzimidazol-2-ylidene] silver(I). Yield: 0.178 g, (79%). m.p.: 237–238 °C; ν(CN): 1454.8 cm−1. Anal. Calc. for C34H50N6O2PdCl2: C: 54.29; H: 6.70; N: 11.17. Found: C: 55.48; H: 6.95; N: 10.83. 1H NMR (300 MHz, CDCI3, δ, ppm); 1.08 (m, 6H, –(CH2)3–CH 3 ); 1.60 (m, 4H, –(CH2)2–CH 2 –CH3); 2.25 (m, 4H, –CH2–CH 2 –C2H5); 2.67 (m, 8H, –NCH 2 CH2O–); 3.25 (m, 4H, –NCH2CH 2 NC4H8O); 3.72 (m, 8H, –NCH2CH 2 O–); 4.88 (t, 4H, J: 7.8 Hz, –CH 2 –C3H7); 5.01 (t, 4H, J: 6.6 Hz, –NCH 2 CH2NC4H8O–); 7.29–7.49 (m, 8H, Ar–H). 13C NMR (75 MHz, CDCI3, δ, ppm); 14 (–(CH2)3–CH3); 20.5 (–(CH2)2–CH2–CH3); 32.2 (–CH2–CH2–C2H5); 48.2 (–NCH 2 CH2O–); 54.2 (–NCH2 CH2NC4H8O); 58.0 (–NCH2CH 2 O–); 58.2 (–CH2–C3H7); 67.4 (–NCH2CH2NC4H8O–); 110.5, 110.8, 111.4, 122.8, 123.7, 134.2 and 134.7 (Ar–C); 181.4 (2-CH).
2.7 General Method for Direct Arylation of Furan and Thiophene with Aryl Bromides
The aryl bromide derivatives (4-bromo acetophenone, 4-bromoanisole and 4-bromo toluene) (1 mmol) and heteroaryl derivatives (2-n-butylfuran and 2-n-butylthiophene) (2 mmol), KOAc (1 mmol) and bis-(NHC)Pd(II) complexes 1a–f (0.003 mmol) were dissolved in N,N-dimethylacetamide (DMAc) (2 mL) in a small Schlenk tube under argon as described in the literature [46]. The reaction mixture was stirred in an oil bath at 130 °C for 1 h then was cooled to room temperature and the solvent was removed under vacuum. The obtained residue was purified by column chromatography (silica gel 60–120 mesh) by using diethyl ether/n-hexane (1:5) as eluent to afford the pure product. The purity of the compounds was checked by gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS). Conversions were calculated by taking into account the conversion of aryl bromides to products.
3 Results and Discussion
3.1 Synthesis of Bis-NHCPd(II) Complexes (1a–f)
The N-benzylbenzimidazole is synthesized by using benzimidazole and benzyl chloride [59, 60] (Fig. 1). The synthetic route for unsymmetrically 2-morpholinoethyl-substituted bis-(NHC)Pd(II) complexes defined in this study was illustrated in Scheme 1. The new bis-(NHC)Pd(II) complexes 1a–f were prepared from the synthesized Ag(I)NHC complexes via transmetallation method. The air and moisture stable the new bis-(NHC)Pd(II) complexes were soluble in solvents such as toluene, dichloromethane, and chloroform. The new bis-(NHC)Pd(II) complexes 1a–f were prepared by mixing chloro[1-alkyl-3-(2-morpholinoethyl) benzimidazol-2-ylidene] silver(I) with 0.5 equivalents of PdCl2(PhCN)2 in dichloromethane (20 mL), then the reaction mixture was stirred at room temperature for 24 h in dark condition. The new bis-(NHC)Pd(II) complexes were obtained as a light yellow solid in 78 to 79% yield. The formations of the nonsymmetrical substituted complexes were confirmed by FT-IR, 1H NMR and 13C NMR spectroscopic methods and elemental analysis techniques. These spectra are consistent with the proposed formulate. In the 13C NMR spectra, the Pd–Ccarbene resonances of this new bis-(NHC)Pd(II) complexes in the 13C NMR spectra appeared highly downfield shifted at δ 181.6, 181.8, 181.8, 182.0, 181.7 and 181.4 ppm for 1a–f, respectively. The results of the elemental analysis, which is one of the analytical techniques used to prove the synthesis of compounds, were evaluated and it was observed that the calculated values were very close to the found values. The FT-IR data clearly indicated the presence of ν(CN) at 1452.6, 1456.7, 1448.1, 1456.7, 1455.2and 1454.8 cm−1 for the new bis-(NHC)Pd(II) complexes (1a–f), respectively. Also, we obtained an appropriate single crystal for complex 1a.

The molecular structure of the N-benzylbenzimidazole, showing the atom labeling. Displacement ellipsoids are drawn at the 25% probability level

Synthesis of bis-(NHC)Pd(II) Complexes 1a–f
3.2 Direct Arylation of 2-n-butylfuran and 2-n-butylthiophene with Various Aryl Bromides
We carried out some experiments for the parameters of direct arylation reaction of para-substituted aryl bromides with 2-n-butylthiophene and 2-n-butylfuran in the presence of 1a–f as the catalyst. The best reaction conditions consisted of at the temperature: 130 °C, base: KOAc, time: 1 h, solvent: DMAc and catalyst loading: 3 mmol% in the literature [46].
Conversions of the products for 2-n-butylthiophene are between 63 and 99% and for 2-n-butylfuran are between 59 and 96% (Tables 2, 3). When 4-bromoacetophenone was used, the best conversion was obtained. However, when we used 4-bromoanisole, it was obtained at low conversion (Tables 2, 3).


Initially, we investigated the binding of 2-n-butylthiophene with 4-bromoacetophenone, 4-bromoanisole and 4-bromotoluene by using complexes 1a–f as the catalyst. When the effects of 1a–f in the formation of the products 2, 3 and 4 were analyzed, conversions of 99–91, 63–89 and 82–93% were observed respectively (Table 2). The use of 4-bromoacetophenone with 2-n-butylthiophene gave the desired coupling product for different complexes (1b, 1d, and 1f) as catalysts in better excellent conversion than the others, such as 95, 99 and 95%, respectively (Table 2). Then, we investigated the binding of 2-n-butylfuran with 4-bromoacetophenone, 4-bromoanisole, and 4-bromotoluene with complexes 1a–f as the catalyst. When the effects of 1a–f in the formation of product 5, 6 and 7 were analyzed, conversions of 86–96, 59–81 and 83–93% were observed respectively (Table 3). The best results were obtained when 2-n-butylthiophene with 4-bromoacetophenone were used (Table 3). Finally, 2-n-butylfuran was bound with 4-bromoanisole to give the arylated products 6 in fewer conversions. When 4-bromoacetophenone and 4-bromotoluene with 2-n-butylfuran were utilized in the direct arylation reaction, conversions of the products (5 and 7) were obtained and observed to be better than the product 6 (Table 3). When compared to similar studies [53, 61], published recently, bis-(NHC)Pd(II) complexes that we have synthesized to appear highly active catalysts.
3.3 Structure Description of the Complex 1a
The results of the single crystal X-ray diffraction analysis illustrate that complex 1a crystallizes with a disordered dichloromethane solvent molecule in the triclinic space group \(P\stackrel{-}{1}\). As depicted in Fig. 2, the asymmetric unit has one-half-molecule and it is completed with a twofold symmetry axis [symmetry code: 1−x, 1−y, 1−z]. The structure shows a distorted square-planar geometry in a trans configuration around the metal center. The Pd–C bond length [2.016 (1) Å] is compatible with the many other trans-Pd(II) complexes, whereas it is longer than the cis-configuration ones [62,63,64,65]. In the study of Huynh et al., these results show that the carbene ligands are more weakly bound to the Pd-center in the trans-form [66]. The planes of the N-benzylbenzimidazole are almost perpendicular [74.65(2)°] to the Pd/C1/C1i/Cl/Cli plane. All other bond lengths and angles are shown in Tables 4 and 5, mostly consistent with the Pd(II) complex studies in the literature [67, 68].

The molecular structure of the complex 1a, showing the atom labeling. Displacement ellipsoids are drawn at the 25% probability level
In the crystal structure, molecules are linked by intermolecular C–H⋯O hydrogen bond [H⋯O = 2.46 Å, C–O = 3.353(2) Å, C–H⋯O = 161°, symmetry code: 1 + x, −1 + y, 1 + z] to form an infinite chain along the ab plane. This hydrogen bond also generates \(R_{2}^{2}(16)\) ring motif (Fig. 3). Moreover, there is a strong C–H⋯pi interaction between the C13 atom of the morpholine ring and the benzene ring of the benzimidazole moiety [Cg: C2/C3/C4/C5/C6/C7; C13–Cg 3.697(3) Å, H13A⋯Cg 2.81 Å, C13–H13A⋯Cg 152°, symmetry code: −x, 1−y, 1−z], which is responsible for the 2D supramolecular network and stabilization of the crystal structure.

Hydrogen bonding of the molecules in the crystal structure and formation of the \(R_{2}^{2}(16)\) ring motif [symmetry codes: (i) 1−x, 1−y, 1−z, (ii) 1 + x, −1 + y, 1 + z, (iii) 2−x, −y, 2−z]
4 Conclusions
As a result, we reported the synthesis of the new 2-morpholinoethyl substituted bis-(NHC)Pd(II) complexes 1a–f. The bis-(NHC)Pd(II) complexes were prepared via the Ag(I)NHC complexes transmetallation route. The catalytic activities of this new 2-morpholinoethyl substituted bis-(NHC)Pd(II) complexes have been examined that they are more efficient and stable catalysts for the direct arylation reactions of 2-n-butylfuran and 2-n-butylthiophene with aryl bromide. The crystal structures determination of the complex 1a and its ligand (N-benzylbenzimidazole) were performed by single crystal X-ray diffraction method. The bis-(NHC)Pd(II) complex 1a has a distorted square planar geometry in the trans configuration. The crystal structure is stabilized by the intermolecular C–H⋯O hydrogen bonds and the C–H⋯pi interaction (Tables 4, 5).
5 Supplementary
Crystallographic data as .cif files for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Center with CCDC 1542124 for complex 1a and 1542126 for N-benzylbenzimidazole. Copies of the data can be obtained free of charge at http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Center, 12, Union Road, Cambridge CB2 1EZ, UK. fax: (+44) 1223-336-033, email: deposit@ccdc.cam.ac.uk.
References
Wanzlick HW, Schönherr HJ (1968) Angew Chem Int Ed Engl 7:141
Öfele K (1968) J Organomet Chem 12:42
Herrmann WA, Weskamp T, Bohm VPW (2001) Adv Organomet Chem 48:3
Arduengo AJ III, Krafczyk R (1998) Chem Unserer Zeit 32:6
Diez Gonzalez S, Nolan SP (2005) Annu Rep Prog Chem B 101:171
Crabtree RH (2006) J Organomet Chem 691:3146
Jafarpour L, Nolan SP (2001) Adv Organomet Chem 46:181
Hu X, Castro-Rodriguez I, Olsen K, Meyer K (2004) Organometallics 23:755
Cavallo L, Correa A, Costabile C, Jacobsen H (2005) J Organomet Chem 690:5407
Kühl O (2009) Coord Chem Rev 253:2481
Garrison JC, Youngs WJ (2005) Chem Rev 105:3978
Winkelmann O, Christian Näther, Lüning UJ (2008) Organomet Chem 693:2784
Viciano M, Mas-Marzá E, Poyatos M, Sanaú M, Crabtree RH, Peris E (2005) Angew Chew Int Ed 44:444
Xi Z, Zhang X, Chen W, Fu S, Wang D (2007) Organometallics 26:6636
Chiu PL, Lee HM (2005) Organometallics 24:1692
Burling S, Field LD, Li HL, Messerle BA, Turner P (2003) Eur J Inorg Chem 17:3179
Bourissou D, Guerret O, Gabbai FP, Bertrand G (2000) Chem Rev 100:39
Berthon-Gelloz G, Markó IE (2006) Wiley, New York, p 119–161
Gade LH, César V, Bellemin-Laponnaz S (2004) Angew Chem Int Ed 43:1014
Diebolt O, Braunstein P, Nolan SP, Cazin CS (2008) J Chem Commun 27:3190
Linninger CS, Herdtweck E, Hoffmann SD, Herrmann WAJ (2008) Mol Struct 890:192
Schneider SK, Herrmann WA, Herdtweck EJ (2006) Mol Catal A 245:248
Altenhoff G, Goddard R, Lehmann CW, Glorius F (2003) Angew Chem Int Ed 42:3690
Organ MG, Avola S, Dubovyk I, Hadei N, Kantchev EAB, O’Brien CJ, Valente C (2006) Chem Eur J 12:4743
Herrmann WA (2002) Angew Chem Int Ed 41:1290
Herrmann WA, Elison M, Fisher J, Kocher C, Artus GRJ (1995) Angew Chem Int Ed 34:2371
Marshall C, Ward MF, Harrison WTA (2004) Tetrahedron Lett 45:5703
Liao CY, Chan KT, Zeng JY, Hu CH, Tu CY, Lee HM (2007) Organometallics 26:1692
Nakamura N, Tajima Y, Sakai K (1982) Heterocycles 17:235
Satoh T, Miura M (2007) Chem Lett 36:200
Bellina F, Cauteruccio S, Rossi R (2008) Curr Org Chem 12:774
McGlacken GP, Bateman LM (2009) Chem Soc Rev 38:2447
Balcells D, Clot E, Eisenstein O (2010) Chem Rev 110:749
Fischmeister C, Doucet H (2011) Green Chem 13:741
Song G, Wang F, Li X (2012) Chem Soc Rev 41:3651
Wencel-Delord J, Glorius F (2013) Nat Chem 5:369
Tsurugi H, Yamamoto K, Nagae H, Kaneko H, Mashima K (2014) Dalton Trans 43:2331
He M, Soulé JF, Doucet H (2014) Chem Cat Chem 6:1824
Yuan K, Soulé JF, Doucet H (2015) ACS Catal 5:978
Negishi E, King AO, Okukado N (1977) J Org Chem 42:1821
Dunina VV, Gorunova ON (2004) Russ Chem Rev 73:309
Bertrand MB, Wolfe JP (2007) Org Lett 9:3073
Sun CL, Li H, Yu DG, Yu M, Zhou X, Lu XY, Huang K, Zheng SF, Li BJ, Shi ZJ (2010) Nat Chem 2:1044
Liu W, Cao H, Lei A (2010) Angew Chem Int Ed 49:2004
Alberico D, Scott ME, Lautens M (2007) Chem Rev 107:174
Özdemir İ, Gök Y, Özeroğlu Ö, Kaloğlu M, Doucet H (2010) Eur J Inorg Chem 12:1798
Aktaş A, Akkoç S, Gök Y (2013) J Coord Chem 66:2901
Aktaş A, Gök Y (2014) Transition Met Chem 39:925
Aktaş A, Gök Y (2015) Catal Lett 145:631
Aktaş A, Gök Y, Akkurt M, Özdemir N (2015) Org Chem Curr Res 4:149
Akkoç S, Gök Y (2014) Appl Organomet Chem 28:854
Gök Y, Akkoç S, Akkurt M, Tahir MN (2014) J Iran Chem Soc 11:1767
Akkoç S, Gök Y (2015) Inorg Chim Acta 429:34
Akkoç S, Gök Y, İlhan IÖ, Kayser V (2016) Beilstein J Org Chem 12:81
Agilent (2014) CrysAlis PRO agilent technologies. Yarnton
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339
Sheldrick GM (2015) Acta Cryst A 71:3
Sheldrick GM (2015) Acta Cryst C 71:3
Vishal K, Fahlman BD, Sasidhar BS, Patil Shivaputra A, Patil Siddappa A (2017) Catal Lett 147(4):900
Chatterjee T, Kumar NT, Das SK (2017) Polyhedron 127:68
Kaloğlu M, Özdemir İ, Dorcet V, Bruneau C, Doucet H (2017) Eur J Inorg Chem 2017:1382
Hahn FE, Holtgrewe C, Pape T (2004) Z Naturforsch 59:1051
Chamizo JA, Morgado J (2000) Transition Met Chem 25:161
Hahn FE, Foth M (1999) J Organomet Chem 585:241
Huynh HV, Neo TC, Tan GK (2006) Organometallics 25:1298
Huynh HV, Ho JHH, Neo TC, Koh LL (2005) J Organomet Chem 690:3854
Salman AW, Rehman GU, Abdullah N, Budagumpi S, Endud S, Abdallah HH, Wong WY (2014) Polyhedron 81:499
Diao Y, Hao R, Kou J, Teng M, Huang G, Chen Y (2013) Appl Organometal Chem 27:546
Acknowledgements
The authors acknowledge İnönü University Scientific and Technology Center for the elemental analyses of the compounds and Dokuz Eylül University for the use of the Rigaku Oxford Xcalibur Eos Diffractometer (purchased under University Research Grant No: 2010.KB.FEN.13).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sarı, Y., Aktaş, A., Barut Celepci, D. et al. Synthesis, Characterization and Crystal Structure of New 2-Morpholinoethyl-Substituted Bis-(NHC)Pd(II) Complexes and the Catalytic Activity in the Direct Arylation Reaction. Catal Lett 147, 2340–2351 (2017). https://doi.org/10.1007/s10562-017-2132-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-017-2132-3