
Abstract
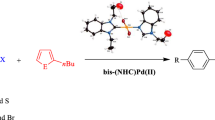
Palladium-catalyzed direct arylation of heteroaromatics has become a popular method for producing carbon–carbon bonds via C–H bond activation. A wide diversity of heteroaromatics such as furan, thiophenes and thiazoles can be used for this reaction. This paper reports the synthesis of N-propylphthalimide-substituted bis-(NHC)PdX2 complexes (NHC = N-heterocyclic carbene), and their catalytic activity in direct arylation reactions. The complexes have been prepared from Ag(I)NHC precursors by transmetallation and characterized by spectroscopy and elemental analysis. The bis-(NHC)PdX2 complexes show excellent activity as catalysts in the direct arylation reactions of 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Direct arylation of aromatic and heteroaromatic C–H bonds has become an important method of aryl C–C bond formation in organic chemistry. Some catalytic systems developed for C–H bond transformations can allow eco-friendly synthesis methods [1,2,3,4,5,6]. One of the most common methods used for the formation of aryl–aryl bonds is by transition metal complex-mediated reactions. Suzuki–Miyaura, Stille and Negishi couplings are among the most important examples of these methods [7, 8].
In recent years, palladium catalytic systems have attracted much attention in metal-catalyzed direct arylation reactions due to their selectivity, efficiency and versatility [9,10,11]. Many chemists have devoted time to the development of new Pd-catalyzed direct arylation reactions. To date, a wide range of electronically rich and poor (hetero)aromatic compounds have been successfully used in palladium-catalyzed direct arylation reactions [12,13,14,15,16,17,18,19], and this approach is one of the most effective methods to access aryl–heteroaryl derivatives [20,21,22].
Numerous studies have been carried out on the catalytic activities of Pd(II)-based complexes containing different ligands [23, 24]. In recent years, Pd–NHC complexes containing N-heterocyclic carbene (NHC) ligands have attracted great interest [25,26,27]. Complexes of NHC ligands have distinctive properties such as being strong σ-donors, weak π-acceptors and good resistance to heat, air and moisture [28,29,30].
In our work, we have investigated the synthesis and characterization of bis-(NHC)PdX2 complexes. Also, we examined the catalytic activities of these complexes in direct arylation reactions. The complexes proved to have fairly good activity, even with low catalyst loadings, in direct arylation reactions with 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole.
Experimental
All synthesis involving bis-(NHC)PdX2 complexes 1a–i were carried out under an inert atmosphere in flame-dried glassware using standard Schlenk techniques. The solvents used were purified by distillation over the drying agents indicated and were transferred under Ar: Et2O (Na/K alloy), CH2Cl2 (P4O10), hexane, toluene (Na). All other reagents were obtained commercially from Merck and Aldrich, and used without further purification. Melting points were measured in glass capillaries under air with an Electrothermal-9200 melting point apparatus. FTIR spectra were obtained in the range of 400–4000 cm−1 on a Perkin–Elmer Spectrum 100 FTIR spectrometer. Proton (1H) and carbon (13C) NMR spectra were recorded using a Varian AS 300 Merkur spectrometer operating at 300 MHz (1H) or 75.47 MHz (13C) in CDCl3 with tetramethylsilane as an internal reference. Reaction products were assayed on an Agilent 6890 N GC system by GC-FID with an HP-5 column of 30 m length, 0.32 mm diameter and 0.25 μm film thickness. Column chromatography was performed using silica gel 60 (70–230 mesh). Elemental analyses were obtained by the İnönü University Scientific and Technological Research Center (Malatya, TURKEY). Crystallographic and physical data of all the complexes are summarized in Table 1.
Preparation of complex 1a
A mixture of bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (0.1g, 0.26 mmol) and bromo[1-methyl-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (0.264 g, 0.52 mmol) in dichloromethane (20 mL) was stirred for 24 h at room temperature in the dark. The mixture was filtered through celite, and the solvents were evaporated under vacuum to afford the product as a white or light yellow solid. The crude product was recrystallized from dichloromethane/diethyl ether (1:3) at room temperature. Yield: 0.21 g (89%). Anal. Calc. for C38H34N6O4PdBr2: C: 50.43, H: 3.79, N: 9.29. Found: C:50.47, H:3.83, N:9.26. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.59 (m, 4H, –(C6H4)NCH2CH 2CH2N–); 4.35 (t, 4H, –(C6H4)NCH2CH2CH 2N– J: 7.5 Hz); 4.44 (s, 6H, –CH 3); 5.01 (m, 4H, –(C6H4)NCH 2CH2CH2N–); 7.29–7.90 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 29.2 (–(C6H4)NCH2 CH2CH2N–); 34.7 (–CH3); 36.1 (–(C6H4)NCH2CH2 CH2N–); 55.6 (–(C6H4)NCH2CH2CH2N–); 110.2, 110.3, 123.1, 123.2, 123.4, 132.1, 132.2, 133.8, 134.1 and 135.1 (Ar–C). 181.5 (2–C–Pd).
Preparation of complex 1b
According to the same procedure as for complex 1a, complex 1b was prepared from bromo[1-ethyl-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (271 mg. 0.52 mmol). Yield: 0.15 g (60%). Anal. Calc. for C40H38N6O4PdBr2: C: 51.49, H: 4.11, N: 9.01. Found: C: 51.46, H: 4.09, N: 9.03. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 1.65 (m, 6H, –CH2CH 3); 2.51 (m, 4H, –(C6H4)NCH2CH 2CH2N–); 3.89 (m, 4H, –(C6H4)NCH2CH2CH 2N–); 4.72 (m, 4H, –CH 2CH3); 4.92 (m, 4H, –(C6H4) NCH 2CH2CH2N–); 7.31–7.90 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 15.2 (–CH2 CH3); 28.8 (–(C6H4)NCH2 CH2CH2N–); 36.2 (–(C6H4)NCH2CH2 CH2N–); 46.2 (–CH2CH3); 55.4 (–(C6H4)NCH2CH2CH2N–); 111.3, 123.5, 132.2, 132.3, 133.7, 133.8, 134.0, 134.1, 134.7 and 134.8 (Ar–C); 181.0 (2–C–Pd).
Preparation of complex 1c
According to the same procedure as for complex 1a, complex 1c was prepared from bromo[1-butyl-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (285 mg. 0.52 mmol). Yield: 86% (221 mg). 0.22 g (86%). Anal. Calc. for C44H46N6O4PdBr2: C: 53.43, H: 4.69, N: 8.50. Found: C: 53.39, H: 4.74, N: 8.47. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 1.09 (t, 3H, –CH2CH2CH2CH 3, J: 6.9 Hz); 1.55 (m, 4H, –CH2CH2CH 2CH3); 2.26 (m, 4H, –CH2CH 2CH2CH3); 2.73 (m, 4H, –(C6H4)NCH2CH 2CH2N–); 3.96 (t, 4H, –(C6H4) NCH2CH2CH 2N, J: 6.9 Hz); 4.81 (m, 4H, –CH 2CH2CH2CH3); 4.93 (m, 4H, –(C6H4)NCH 2CH2CH2N–); 7.27–7.89 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 13.7 (–CH2CH2CH2 CH3); 20.4 (–CH2CH2 CH2CH3); 29.4 (–(C6H4)NCH2 CH2CH2N–); 35.1 (–(C6H4)NCH2CH2 CH2N–); 41.0 (–CH2 CH2CH2CH3); 56.1 (–CH2CH2CH2CH3); 56.6 (–(C6H4)NCH2CH2CH2N–); 110.9, 111.2, 123.0, 123.3, 123.4, 132.1, 133.1 and 134.1 (Ar–C); 181.2 (2–C–Pd).
Preparation of complex 1d
According to the same procedure as for complex 1a, complex 1d was prepared from bromo[1-(2-methoxyethyl)-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (287 mg. 0.52 mmol). Yield: 0.19 g (75%). Anal. Calc. for C42H42N6O6PdBr2: C: 50.80, H: 4.26, N: 8.46. Found: C: 50.73, H: 5.22, N: 8.42. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 3.26 (s, 6H, –CH2CH2OCH 3); 3.90 (m, 4H, –CH2CH 2OCH3); 4.97 (m, 4H, –CH 2CH2OCH 3); 2.51 (m, 4H, –(C6H4)NCH2CH 2CH2N); 4.26 (m, 4H, –(C6H4)NCH2CH2CH 2N); 4.99 (m, 4H, –(C6H4)NCH 2CH2CH2N); 7.37–7.91 (m, 16H, Ar–H).13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 29.4 (–(C6H4)NCH2 CH2CH2N); 35.2 (–(C6H4)NCH2CH2 CH2N); 56.6 (–(C6H4)NCH2CH2CH2N); 29.1 (–CH2CH2OCH3); 36.2 (–CH2 CH2OCH3); 58.9 (–CH2CH2OCH3); 123.4, 123.5, 132.2, 132.3, 134.7 and 134.8 (Ar–C); 181.9 (2–C–Pd).
Preparation of complex 1e
According to the same procedure as for complex 1a, complex 1e was prepared from bromo[1-(2-ethoxyethyl)-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (294 mg. 0.52 mmol). Yield: 0.17 g (65%). Anal. Calc. for C44H46N6O6PdBr2: C: 51.76, H: 4.54, N: 8.23. Found: C: 51.87, H: 4.64, N: 8.27. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 1.24 (t, 6H, –CH2CH2OCH2CH 3, J: 7.2 Hz); 2.64 (m 4H, –(C6H4)NCH2CH 2CH2N–); 3.50 (m 4H, –CH2CH2OCH 2CH3); 4.12 (m, 4H, –(C6H4) NCH2CH2CH 2N–); 4.14 (t, 4H, –CH2CH 2OCH2CH3, J: 7.2 Hz); 4.71 (m, 4H, –CH 2CH2OCH2CH3); 5.15 (m, 4H, –(C6H4)NCH 2CH2CH2N–); 7.28–7.91 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 15.2 (–CH2CH2OCH2 CH3); 28.9 (–(C6H4)NCH2 CH2CH2N–); 36.3 (–(C6H4)NCH2CH2 CH2N–); 46.2 (–CH2CH2OCH2CH3); 66.8 (–(C6H4)NCH2CH2CH2N–); 69.8 (–CH2 CH2OCH2CH3); 70.2 (–CH2CH2OCH2CH3); 109.9, 110.2, 111.6, 112.1, 123.2, 123.3, 132.1, 132.2, 133.8 and 134.1 (Ar–C); 181.6 (2–C–Pd).
Preparation of complex 1f
According to the same procedure as for complex 1a, complex 1f was prepared from chloro[1-(3-methylbenzyl)-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (288 mg. 0.52 mmol). Yield: 0.21 g (81%). Anal. Calc. for C52H46N6O4PdCl2: C: 62.69, H: 4.65, N: 8.44. Found: C: 62.65, H: 4.61, N: 8.38. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.32 (s, 6H, –CH2C6H4(CH 3)); 2.81 (m, 4H, –(C6H4)NCH2CH 2CH2N–); 4.19 (t, 4H, –(C6H4)NCH2CH2CH 2N, J: 7.2 Hz); 5.14 (t, 4H, –(C6H4)NCH 2CH2CH2N, J: 7.2 Hz); 5.97 (s, 4H, –CH 2C6H4(CH3)); 6.99–7.83 (m, 24H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 21.3 (–CH2C6H4(CH3)); 29.0 (–(C6H4)NCH2 CH2CH2N); 36.3 (–(C6H4)NCH2CH2 CH2N); 46.3 (–(C6H4)NCH2CH2CH2N); 52.7 (–CH2C6H4(CH3)); 110.2, 111.4, 123.1, 124.7, 124.9, 128.4, 128.7, 132.1, 133.9, 134.2, 134.3, 134.5, 135.4, 135.6, 138.3 and 138.6 (Ar–C); 168.2 (C=O); 182.1(2–C–Pd).
Preparation of complex 1g
According to the same procedure as for complex 1a, complex 1g was prepared from chloro[1-(4-methylbenzyl)-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (288 mg. 0.52 mmol). Yield: 0.20 g (78%). Anal. Calc. for C52H46N6O4PdCl2: C: 62.69, H: 4.65, N: 8.44. Found: C: 62.75, H: 4.69, N: 8.48. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.27 (s, 6H, –CH2C6H4(CH 3 )); 2.59 (m, 4H, –(C6H4)CH2CH 2CH2N–); 3.71 (t, 4H, –(C6H4)CH2CH2CH 2N, J: 7.2 Hz); 4.91 (t, 4H, –(C6H4)CH 2CH2CH2N–, J: 7.2 Hz); 5.96 (s, 4H, –(C6H4)CH 2C6H4(CH3)); 7.02–7.86 (m, 24H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 21.1 (–CH2C6H4(CH3)); 28.9 (–(C6H4)CH2 CH2CH2N–); 36.3 (–(C6H4)CH2CH2 CH2N–); 46.1 (–(C6H4)CH2CH2CH2N–); 52.3 (–CH2C6H4(CH3)); 110.2, 110.5, 111.5, 123.0, 123.3, 127.6, 127.8, 129.3, 132.1, 132.5, 132.7, 133.9, 134.2, 134.3, 134.5, 137.3 ve 137.5 (Ar–C); 167.9 and 168.2 (C=O); 181.9 (2–C–Pd).
Preparation of complex 1h
According to the same procedure as for complex 1a, complex 1h was prepared from chloro[1-(2,4,6-trimethylbenzyl)-3-(N-propylphthalimide)benzimidazol-2-ylidene]silver(I) (302 mg. 0.52 mmol). Yield: 0.23 g (87%). Anal. Calc. for C56H54N6O4PdCl2: C: 63.91, H: 5.17, N: 7.99. Found: C: 64.01, H: 5.20, N: 8.04. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.77 (m, 4H, –(C6H4)CH2CH 2CH2N–); 4.16 (t, 4H, –(C6H4)CH2CH2CH 2N–, J: 7.5 Hz); 5.23 (t, 4H, –(C6H4)CH 2CH2CH2N–, J: 7.5 Hz); 2.27 and 2.35 (s, 18H,CH2C6H2(CH 3)3); 6.18 (s, 4H, –CH 2C6H2(CH3)3), 6.38–7.82 (m, 20H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 20.9 and 21.1 (–CH2C6H2(CH3)3); 29.0 (–(C6H4)CH2 CH2CH2N); 36.3 (–(C6H4)CH2CH2 CH2N); 46.3 (–(C6H4)CH2CH2CH2N); 49.6 (–CH2C6H2(CH3)3); 110.3, 111.8, 122.8, 123.2, 128.2, 129.6, 132.0, 132.2, 133.8, 134.3, 134.5, 138.2, 138.5 ve 138.9 (Ar–C); 168.1 (C=O); 182.0 (C–Pd).
Preparation of complex 1i
According to the same procedure as for complex 1a, complex 1i was prepared from chloro[1-naftalenomethyl-3-(N-propylphthalimide)benzimidazol-2-ylidene] silver(I) (306 mg. 0.52 mmol). Yield: 0.23 g (84%). Anal. Calc. for C58H46N6O4PdCl2: C: 65.21, H: 4.34, N: 7.87. Found: C: 65.14, H: 4.32, N: 7.85. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.83 (m, 4H, (–(C6H4)CH2CH 2CH2N–); 4.21 (t, 4H, –(C6H4)CH2CH2CH 2N–, J: 7.2 Hz); 5.18 (t, 4H, –(C6H4)CH 2CH2CH2N–, J: 7.2 Hz); 6.19 (s, 4H, –CH 2C10H7); 6.72–7.88 (m, 30H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 29.1 (–(C6H4)CH2 CH2CH2N–); 36.3 (–(C6H4)CH2CH2 CH2N–); 46.3 (–(C6H4)CH2CH2CH2N–); 49.5 (CH2C10H7); 110.4, 111.3, 122.3, 123.2, 125.2, 125.9, 126.2, 126.8, 128.3, 128.9, 130.3, 130.9, 132.3, 133.1, 133.9, 134.7 ve 134.8 (Ar–C); 168.3 (C=O); 182.6 (2–C–Pd).
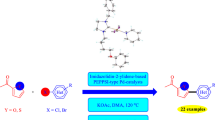
Procedure for arylation of furan, thiophene and thiazole
The heteroaryl derivatives (2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole) (2 mmol), the aryl bromide derivatives (4-bromo acetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene) (1 mmol), KOAc (1 mmol) and bis-(NHC)PdX2 complexes 1a–i (0.003 mmol) were dissolved in N,N-dimethylacetamide (DMAc) (2 mL) in a small Schlenk tube under argon as described in the literature [31]. The mixture was stirred in an oil bath at 130 °C for 1 h, then cooled to room temperature, and the solvent was removed under vacuum. The residue was purified by column chromatography (silica gel 60–120 mesh) using diethyl ether/n-hexane (1:5) as eluent to afford the pure product. The purities of the compounds were checked by GC and GC–MS. Conversions were calculated based on the aryl bromide.
Results and discussion
Synthesis of bis-(NHC)PdX2 parent complexes (1a–i)
The synthetic route for the N-propylphthalimide substituted bis-(NHC)PdX2 complexes is illustrated in Scheme 1. The bis-(NHC)PdX2 complexes 1a-i were prepared from the corresponding N-propylphthalimide substituted Ag(I)NHC complexes via transmetallation, as reported in the literature [32]. The bis-(NHC)PdX2 complexes were obtained as light yellow solids in 60–89% yields. Resistant to air and moisture, the complexes are soluble in solvents such as DMF and DMSO, but less soluble in halogenated solvents such as chloroform and dichloromethane. Formation of the N-propylphthalimide substituted complexes was confirmed by FTIR, 1H NMR and 13C NMR spectroscopy and by elemental analysis. The 13C NMR spectra of the complexes reveal the Pd–C(carbene) signal as a singlet between at 181.0 and 182.6 ppm [33], instead of the Ag–C(carbene) singlet observed between 188 and 190 ppm for the Ag(I)NHC complexes [34], confirming the successful transmetallation reaction. The results of the elemental analysis were in good agreement with the theoretical values. The FTIR spectra of all of the complexes 1a–i show a strong band at 1426–1465 cm−1 for ν(CN) (Table 1). Unfortunately, despite all our efforts, we could not obtain a single crystal from these new complexes for X-ray diffraction studies.

Synthesis of N-propylphthalimide substituted bis-(NHC)PdX2 complexes 1a–i. X = Cl or Br

The direct arylation reaction of 2-n-butylfuran with aryl bromides by complexes 1a–i

The direct arylation reaction of 2-n-butylthiophene with aryl bromides by complexes 1a–i

The direct arylation reaction of 2-isopropylthiazole with aryl bromides by complexes 1a–e
Direct arylation of 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole
We have investigated the direct arylation of para-substituted aryl bromides with 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole in the presence of 1a–i as catalyst. Product conversions for 2-n-butylfuran were between 60 and 99%, for 2-n-butylthiophene between 51 and 99%, and for 2-isopropylthiazole between 71 and 99% (Tables 2, 3 and 4).
Initially, we investigated the reactions of 2-n-butylfuran with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene using complexes 1a–i as catalysts. Conversions of 77–97%, 60–96%, 61–99% and 70–85% were observed for R = –COCH3, –OCH3, –CH3 and –H, respectively (Table 2). Generally, the conversions for substrates containing electron-withdrawing groups were higher than those for substituents containing electron-donating groups.
Next, we investigated the reactions of 2-n-butylthiophene with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene using complexes 1a–i as catalysts. Conversions of 92–99%, 51–81%, 61–94% and 91–99% were observed R=–COCH3, –OCH3, –CH3 and –H, respectively (Table 3).
Finally, we investigated the reactions of 2-isopropylthiazole with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene using complexes 1a–e as catalysts. Conversions of 96–99%, 71–92%, 84–99% and 73–96% were observed R = –COCH3, –OCH3, –CH3 and –H, respectively (Table 4). When compared to similar studies [32, 35] published recently, the bis-(NHC)PdX2 complexes that we have synthesized in this work appear to be highly active catalysts.
Conclusions
We have reported the synthesis of N-propylphthalimide substituted bis-(NHC)PdX2 complexes from the corresponding Ag(I)NHC complexes via transmetallation. The catalytic activities of these N-propylphthalimide substituted bis-(NHC)PdX2 complexes show that they are efficient and stable catalysts for the direct arylation reactions of 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole with aryl bromides.
References
Kakiuchi F, Murai S (2002) Acc Chem Res 35:826
Miura M, Nomura M (2002) Top Curr Chem 219:211
Kakiuchi F, Chatani N (2003) Adv Synth Catal 345:1077
Alberico D, Scott ME, Lautens M (2007) Chem Rev 107:174
Sun L, Li H, Yu D-G, Yu M, Zhou X, Lu X-Y, Huang K, Zheng S-F, Li B-J, Shi Z-J (2010) Nat Chem 2:1044
Liu W, Cao H, Lei A (2010) Angew Chem Int Ed 49:2004
Stanforth SP (1998) Tetrahedron 54:263
Hassan M, Se’vignon M, Gozzi C, Shulz E, Lemaire M (2002) Chem Rev 102:1359
Miura M, Nomura M (2002) Top Curr Chem 219:211
Kakiuchi F, Chatani N (2003) Adv Synth Catal 345:1077
Alberico D, Scott ME, Lautens M (2007) Chem Rev 107:174
Yokooji A, Satoh T, Miura M, Nomura M (2004) Tetrahedron 60:6757
Campeau L-C, Rousseaux S, Fagnou K (2005) J Am Chem Soc 127:18020
Do H-Q, Khan RMK, Daugulis O (2008) J Am Chem Soc 130:15185
Liégault B, Lapointe D, Caron L, Vlassova A, Fagnou KJ (2009) Org Chem 74:1826
Ueda K, Yanagisawa S, Yamaguchi J, Itami K (2010) Angew Chem Int Ed 49:8946
Tamba S, Fuji R, Mori A, Hara K, Koumura N (2011) Chem Lett 40:922
Fu HY, Chen L, Doucet H (2012) J Org Chem 77:4473
Reddy VP, Qiu R, Iwasaki T, Kambe N (2013) Org Lett 15:1290
Bheeter CB, Chen L, Soulé J-F, Doucet H (2016) Catal Sci Technol 6:2005
Yuan K, Doucet H (2013) Chem Cat Chem 5:3495
Tsurugi H, Yamamoto K, Nagae H, Kaneko H, Mashima K (2014) Dalton Trans 43:2331
Khake SM, Jagtap RA, Dangat YB, Gonnade RG, Vanka K, Punji B (2016) Organometallics 35:875
Abdellaoui F, Youssef C, Ammar HB, Roisnel T, Soulé J-F, Doucet H (2016) ACS Catal 6:4248
Akkoç S, Gök Y (2015) Inorg Chim Acta 429:34
Yiğit M, Yiğit B, Gök Y (2016) Inorg Chim Acta 453:23
Lin N-C, Syu HJH, Naziruddin AR, Liu F-C, Lin IJB (2017) RSC Adv 7:11652
Herrmann WA (2002) Angew Chem Int Ed 41:1290
Peris E, Crabtree RH (2004) Coord Chem Rev 248:2239
Burling S, Whittlesey MK, Williams JMJ (2005) Adv Synth Catal 347:591
Zhang CM, Trudell ML (2000) Tetrahedron Lett 41:595
Akkoç S, Gök Y (2014) Appl Organometal Chem 28:854
Sarı Y, Aktaş A, Barut Celepci D, Gök Y, Aygün M (2017) Catal Lett 147:2340
Gök Y, Akkoç S, Erdoğan H, Albayrak S (2016) J Enzym Inhib Med Chem 31:1322
Yiğit M, Yiğit B, Gök Y (2016) Inorg Chim Acta 453:23
Acknowledgements
This work was financially supported by İnönü University Research Fund (IUBAP 2012/02) and the authors acknowledge İnönü University Scientific and Technology Center for the elemental analyses of the compounds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Erdoğan, H., Aktaş, A., Gök, Y. et al. N-Propylphthalimide-substituted bis-(NHC)PdX2 complexes: synthesis, characterization and catalytic activity in direct arylation reactions. Transit Met Chem 43, 31–37 (2018). https://doi.org/10.1007/s11243-017-0190-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0190-4