
Abstract
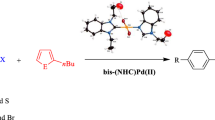
Almost in all fields of chemistry, the C–C cross-coupling reactions such as the direct arylation reaction have attracted much attention and have advanced quickly in recent years due to the importance of environment-friendly properties. This study contains the synthesis of the 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes and their catalytic activity in direct arylation reaction. The bis(NHC)PdX2 complexes have been prepared from the 2-methyl-1,4-benzodioxan-substituted Ag(I)NHC complexes via transmetallation method. The bis(NHC)PdX2 complexes have been characterized using 1H NMR, 13C NMR, FT-IR spectroscopy and elemental analysis techniques. The bis(NHC)PdX2 complexes have been examined as catalysts in the direct arylation reactions with 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole, and have demonstrated excellent activity in these reactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The importance of NHC ligands in organic and organometallic chemistry is increasing day by day [1,2,3,4]. It is emphasized that the remarkable effect of NHC ligands on transition metal catalysis is because of the strong sigma donor character even in the oldest reviews [5,6,7]. It is believed that in the cross-coupling reactions, in particular, the strong M–NHC bond prevents the decomposition of the molecular catalysts [8], and stabilizes the higher oxidation states necessary in multiple catalytic contexts.
Metal–NHC complexes are unique organometallic compounds in which numerous studies have been carried out on both catalytic activity studies and medical applications. The NHC–palladium complexes are simply synthesized by means of different NHC salts with Pd(OAc)2 [9,10,11,12] and using the transmetallation method [13,14,15] in which Ag–NHC complexes are used as the transfer agent with PdCl2(PhCN)2 [16] or PdCl2(CH3CN)2 [17, 18]. From these complexes, medical applications [19,20,21] of Pd(II)NHC complexes are known, but Pd(II)NHC complexes are more well-known for their unique catalytic properties [22,23,24].
In recent years, formation of carbon–carbon bonds via C–H bond activation has become a popular method for palladium-catalyzed arylation of heteroaromatics. For this reaction, most of the heteroaromatic compounds such as (benzo)thiophenes, (benzo)furans, pyrroles, indoles, oxazoles, imidazoles, pyrazoles or triazoles can be used [25]. In recent years important studies have been published on the catalytic properties of palladium-based complexes [26,27,28,29,30]. In particular, the catalytic activities on the direct arylation reactions of the palladium-based complexes containing NHC ligands are becoming more and more important [31,32,33,34,35,36,37,38]. Therefore, we researched the synthesis and the characterization of the 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes. Also, we investigated the catalytic activities of the 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes in direct arylation reactions, and they demonstrated quite good activity as catalysts in the direct arylation reactions with 2-n-butylfuran, 2-n-butylthiophene, and 2-isopropylthiazole.
Experimental
All synthesis involving bis(NHC)PdX2 complexes 1a–h were carried out under an inert atmosphere in flame-dried glassware using standard Schlenk techniques. The solvents used were purified by distillation over the drying agents indicated and were transferred under Ar: Et2O (Na/K alloy), CH2Cl2 (P4O10), hexane, toluene (Na).
All other reagents were commercially available from Merck and Aldrich Chemical Co. and used without further purification. Melting points were identified in glass capillaries under air with an Electrothermal-9200 melting point apparatus. FT-IR spectra were saved in the range 400–4000 cm−1 on PerkinElmer Spectrum 100 FT-IR spectrometer. Proton (1H) and Carbon (13C) NMR spectra were recorded using either a Varian AS 300 Merkur spectrometer operating at 300 MHz (1H), 75.47 MHz (13C) in CDCl3 with tetramethylsilane as an internal reference. All reactions were observed on an Agilent 6890 N GC system by GC-FID with an HP-5 column of 30 m length, 0.32 mm diameter and 0.25 µm film thickness. Column chromatography was performed using silica gel 60 (70–230 mesh). Elemental analyses were performed by İnönü University Scientific and Technological Research Center (Malatya, TURKEY).
Synthesis
Synthesis of dibromo-bis[1-methyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]palladium(II), 1a
Bromo[1-methyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I)) (234 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol) in dichloromethane (20 mL) were stirred for 24 h at room temperature in dark conditions. Then, they were filtered through celite and the solvents were evaporated under vacuum to make the product into a white or light yellow solid. The crude product was recrystallized from dichloromethane/diethyl ether (1:3) at room temperature. Yield: 84% (174 mg). Anal. Calc. for C34H32N4O4PdBr2: C: 49.39, H: 3.90, N: 6.78. Found: C: 49.47, H: 4.83, N: 6.72. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 4.44 (m, 4H, –NCH2CHOCH2–); 4.46 (s, 6H, –CH3); 5.28 (m, 4H, –OCHCH2O–); 5.38 (m, 2H, –CH2CHOCH2–); 6.93–7.54 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO–d6) δ (ppm) = 34.5 (–CH3); 47.8 (OCH2CH(CH2)O); 53.5 (OCH2CH(CH2)O); 72.6 (OCH2CH(CH2)O); 117.5, 117.6, 117.7, 121.7, 122.2, 123.5, 134.9 and 143.1(Ar–C); 181.8 (2-C–Pd).
Synthesis of dibromo-bis[1-butyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]palladium(II), 1b
Using the same method for the synthesis of complex 1a, the 1b complex was prepared from bromo[1-butyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (255 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 79% (180 mg). Anal. Calc. for C40H44N4O4PdBr2: C: 52.73, H: 4.87, N: 6.15. Found: C: 52.68, H: 4.82, N: 6.11. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 0.97 (t, 6H, –CH2CH2CH2CH3, J: 6.8 Hz); 1.53 (m, 4H, –CH2CH2CH2CH3); 2.31 (m, 4H, –CH2CH2CH2CH3); 4.37 (m, 4H, –NCH2CHOCH2–); 5.23 (t, 4H, –NCH2CH2CH2CH3J: 6.0 Hz); 5.34 (m, 2H, –CH2CHOCH2–); 5.38 (d, 4H, –OCHCH2O– J: 4.6 Hz); 7.05–7.49 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 14.3 (–CH2CH2CH2CH3); 20.3 (–CH2CH2CH2CH3); 32.4 (–CH2CH2CH2CH3); 47.3 (OCH2CH(CH2)O); 51.3 (–CH2CH2CH2CH3); 53.2 (OCH2CH(CH2)O); 72.4 (OCH2CH(CH2)O); 117.2, 117.4, 117.5, 121.4, 122.0, 123.3, 135.0 (Ar–C); 181.6 (2-C–Pd).
Synthesis of dibromo-bis[1-(2-methoxyethyl)-3-(2-methyl-1,4-benzodioxan) benzimidazol-2-ylidene]palladium(II), 1c
Using the same method for the synthesis of complex 1a, the 1c complex was prepared from bromo[1-(2-methoxyethyl)-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (256 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 87%. (200 mg). Anal. Calc. for C38H40N4O6PdBr2: C: 49.88, H: 4.41, N: 6.12. Found: C: 49.91, H: 4.42, N: 6.14. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 3.38 (s, 6H, –CH2CH2OCH3); 4.32 (t, 4H, –CH2CH2OCH3J: 7.2 Hz); 4.39 (m, 4H, –NCH2CHOCH2–); 4.42 (m, 4H, –CH2CH2OCH3J: 7.2 Hz); 5.40 (d, 4H, –OCHCH2O–J: 4.6 Hz); 5.44 (m, 2H, –CH2CHOCH2–); 6.90–7.04 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 28.6 (–CH2CH2OCH3); 35.9 (–CH2CH2OCH3); 47.5 (OCH2CH(CH2)O); 53.4 (–CH2CH2OCH3); 58.5 (OCH2CH(CH2)O); 72.5 (OCH2CH(CH2)O); 117.5, 117.6, 117.7, 121.7, 122.1, 123.4, 134.7, 135.1, 142.3 and 143.2 2 (Ar–C); 182.4 (2-C–Pd).
Synthesis of dibromo-bis[1-(2-ethoxyethyl)-3-(2-methyl-1,4-benzodioxan) benzimidazol-2-ylidene]palladium(II), 1d
Using the same method for the synthesis of complex 1a, the 1d complex was prepared from bromo[1-(2-ethoxyethyl)-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (263 mg 0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 81% (234 mg). Anal. Calc. for C40H44N4O6PdBr2: C: 50.95, H: 4.70, N: 5.95. Found: C: 50.89, H: 4.68, N: 6.01. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 1.06 (t, 6H, –CH2CH2OCH2CH3, J: 6.9 Hz); 3.48 (m 4H, –CH2CH2OCH2CH3); 4.13 (m, 4H, –CH2CH2OCH2CH3); 4.48 (m, 4H, –NCH2CHOCH2–); 4.71 (m, 4H, –CH2CH2OCH2CH3); 5.20 (d, 4H, –OCHCH2O– J: 4.6 Hz); 5.40 (m, 2H, –CH2CHOCH2–); 6.82–7.78 (m, 16H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 15.3 (–CH2CH2OCH2CH3); 35.9 (–CH2CH2OCH2CH3); 47.5 (OCH2CH(CH2)O); 53.6 (–CH2CH2OCH2CH3); 66.3 (OCH2CH(CH2)O); 72.3 (–CH2CH2OCH2CH3); 73.2 (OCH2CH(CH2)O); 111.7, 112.2, 117.6, 117.8, 122.1, 122.3, 123.7, 134.8, 142.7 and 143.3 (Ar–C); 182.0 (2-C–Pd).
Synthesis of dichloro-bis[1-benzyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]palladium(II), 1e
Using the same method for the synthesis of complex 1a, the 1e complex was prepared from chloro[1-benzyl-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (250 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 86% (190 mg). Anal. Calc. for C46H40N4O4PdCl2: C: 62.07, H: 4.53, N: 6.29. Found: C: 62.11, H: 4.56, N: 6.32. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 4.05 (d, 4H, –NCH2CHOCH2– J: 9.9 Hz); 4.57 (d, 4H, –OCHCH2O– J: 9.9 Hz); 4.93 (m, 2H, –CH2CHOCH2–); 5.21 (s, 4H, –NCH2C6H5); 6.68–7.67 (m, 26H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 48.1 (OCH2CH(CH2)O); 52.5 (–CH2C6H5); 65.1 (OCH2CH(CH2)O); 72.6 (OCH2CH(CH2)O); 111.1, 117.3, 117.7, 121.5, 121.9, 122.2, 123.5, 127.4, 127.8, 128.2, 128.7, 129.1, 133.9, 134.1, 135.2, 135.4, 142.3 and 143.2 (Ar–C); 182.5(2-C–Pd).
Synthesis of dichloro-bis[1-(4-methylbenzyl)-3-(2-methyl-1,4-benzodioxan) benzimidazol-2-ylidene]palladium(II), 1f
Using the same method for the synthesis of complex 1a, the 1f complex was prepared from chloro[1-(4-methylbenzyl)-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (257 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 90% (207 mg). Anal. Calc. for C48H44N4O4PdCl2: C: 62.79, H: 4.83, N: 6.10. Found: C: 62.82, H: 4.79, N: 6.12. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.29 (s, 6H, –CH2C6H4(CH3)); 4.06 (d, 4H, –NCH2CHOCH2– J: 10.8 Hz); 4.52 (m, 2H, –CH2CHOCH2–); 4.96 (d, 4H, –OCHCH2O– J: 10.8 Hz); 5.33 (s, 4H, –NCH2C6H4(CH3); 6.32–7.59 (m, 24H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 21.2 (–CH2C6H4(CH3)); 48.1 (OCH2CH(CH2)O); 53.3 (–CH2C6H4(CH3)); 65.8 (OCH2CH(CH2)O); 72.8 (OCH2CH(CH2)O); 111.1, 117.3 117.7, 121.5, 121.7, 122.1, 123.3, 129.4, 129.7, 132.2, 133.9, 134.2, 135.4, 137.5, 142.2 and 143.2 (Ar–C); 182.3 (2-C–Pd).
Synthesis of dichloro-bis[1-(2,4,6-trimethylbenzyl)-3-(2-methyl-1,4-benzodioxan) benzimidazol-2-ylidene]palladium(II), 1g
Using the same method for the synthesis of complex 1a, the 1g complex was prepared from chloro[1-(2,4,6-trimethylbenzyl)-3-(2-methyl-1,4-benzodioxan)benzimidazol-2-ylidene]silver(I) (271 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 80% (195 mg). Anal. Calc. for C52H52N4O4PdCl2: C: 64.10, H: 5.38, N: 5.75. Found: C: 64.08, H: 5.35, N: 5.77. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 2.31 and 2.41 (s, 18H, –CH2C6H2(CH3)3); 4.50 (d, 4H, –OCHCH2O– J: 6.6 Hz); 5.23 (d, 4H, –NCH2CHOCH2– J: 9.9 Hz); 5.32 (s, 4H, –CH2C6H2(CH3)3), 5.48 (m, 2H, –CH2CHOCH2–); 6.03–7.51 (m, 20H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 21.0 and 21.2 (–CH2C6H2(CH3)3); 47.9 (OCH2CH(CH2)O); 51.1 (–CH2C6H2(CH3)3); 65.1 (OCH2CH(CH2)O); 72.3 (OCH2CH(CH2)O); 110.4, 111.7, 112.1, 117.3, 117.7, 121.5, 121.9, 122.2, 122.9, 123.3, 127.8, 129.7, 134.5, 135.3, 138.6, 138.8, 142.3 and 143.2 (Ar–C); 182.4 (C–Pd).
Synthesis of dichloro-bis[1-(2-methyl-1,4-benzodioxan)-3-(4-vinylbenzyl)benzimidazol-2-ylidene] palladium(II), 1h
Using the same method for the synthesis of complex 1a, the 1h complex was prepared from chloro[1-(2-methyl-1,4-benzodioxan)-3-(4-vinylbenzyl)benzimidazol-2-ylidene] silver(I) (263 mg·0.50 mmol) and bis(benzonitrile)palladium(II) chloride [PdCl2(PhCN)2] (95 mg·0.25 mmol). Yield: 84% (198 mg). Anal. Calc. for C50H44N4O4PdCl2: C: 63.74, H: 4.71, N: 5.95. Found: C: 63.76, H: 4.74, N: 5.92. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 4.07 (d, 4H, –NCH2CHOCH2– J: 7.2 Hz); 4.32 (m, 2H, –CH2CHOCH2–); 4.57 (d, 4H, –OCHCH2O– J: 7.2 Hz); 5.23 and 5.75 (d, 4H, CH2C6H4CH = CH2, J: 7.5 Hz); 5.84 (s, 4H, CH2C6H4CH = CH2); 6.60 (dd, 2H, C6H4CH = CH2, J: 7.5 Hz); 6.74–7.67 (m, 24H, Ar–H). 13C NMR (75.47 MHz, DMSO-d6) δ (ppm) = 48.3 (OCH2CH(CH2)O); 51.4 (–CH2C6H4CH = CH2); 65.4 (OCH2CH(CH2)O); 71.6 (OCH2CH(CH2)O); 111.3, 113.8, 115.2, 117.5, 117.8, 123.5, 126.6, 126.8, 127.1, 127.8, 128.2, 128.7, 131.1, 132.2, 135.9, 136.3 and 138.4 (Ar–C and –CH=CH2); 182.1 (2-C–Pd).
Results and discussion
Synthesis of bis(NHC)PdX2 synthetic route complexes (1a–h)
The parent 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes 1a–h were synthesized using the common silver carbene transfer method. The silver complexes were directly used for transmetalation to palladium(II) in a 2/1 ratio to afford the desired bis(NHC)PdX2 complexes which have been illustrated in Scheme 1. The bis(NHC)PdX2 complexes 1a–h were prepared by mixing [PdCl2(PhCN)2] with 0.5 equivalents of chloro(or bromo)[1-alkyl(or aryl)-3-(2-methyl-1,4-benzodioxan) benzimidazol-2-ylidene]silver(I) in dichloromethane (20 mL), then the reaction mixture was stirred at room temperature for 24 h in dark conditions. The bis(NHC)PdX2 complexes were obtained as a light yellow and white solid in 79–89% yield. The air- and moisture-stable bis(NHC)PdX2 complexes were soluble in solvents such as dimethylformamide and dimethylsulfoxide. The bis(NHC)PdX2 complexes were less soluble in halogens such as chloroform and dichloromethane. The formation of the 2-methyl-1,4-benzodioxan-substituted complexes was confirmed by FT-IR, 1H NMR and 13C NMR spectroscopic methods and elemental analysis techniques. These spectra are consistent with the proposed formulate. This condition indicated the successful transmetallation and confirmed the formation of the expected bis(NHC)PdX2 complexes. In the 13C NMR spectra, the Pd-Ccarbene resonances of this bis(NHC)PdX2 complexes in the 13C NMR spectra appeared highly downfield when it shifted at δ 181.8, 181.60, 181.4, 182.0, 182.5, 182.3, 182.4, and 182.1 ppm for 1a–h, respectively. The results of the elemental analysis, which is one of the most analytical techniques used to prove the synthesis of compounds, were evaluated and it was observed that the calculated values were very close to the found values. The FT-IR data clearly indicated the presence of ν(CN) at 1452, 1444, 1455, 1454, 1445, 1447, 1445, and 1447 cm−1 for the bis(NHC)PdX2 complexes (1a–i), respectively. Crystallographic and physical data of all compounds are summarized in Table 1.

Synthesis of 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes 1a–h
Proposed catalytic cycle for bis(NHC)PdX2 complexes
The proposed general catalytic pathway is shown in Scheme 2. In this cycle, first the oxidative addition of the aryl halide to the pre-activated bis(NHC)PdX2 complex takes place. It is believed that oxidative addition occurs more readily in the presence of the NHC complex thanks to strong sigma donor properties [39, 40]. Then, the ligand exchange occurs with the effect of the base. The character of the base in this step has important due to both oxidative addition and reductive elimination. Then, the 2-substituted furan/thiophene molecule is added to the complex. In this step, activation of C–H occurs at position 5. In the course of C–H activation, the NHC ligand contributes to stabilizing of the occurring complex. Lastly, C5-arylated furan/thiophene product has been obtained by the result of reductive elimination. In this step, the steric bulk of the bis(NHC)PdX2 complex facilitates elimination of arylated furan/thiophene product.

Proposed catalytic cycle for the direct C–H bond arylation for bis(NHC)PdX2 complexes
In the bis(NHC)PdX2 complexes, the electronic properties provided by the alkyl substituents attached to the N atom provide the stability of the complex in the catalytic cycle, while the steric bulk of the ligand facilitating reductive elimination in the catalytic cycle. Therefore, we used ligands with different electronic and steric properties on the bis(NHC)PdX2 complexes.
General method for direct arylation of furan and thiophene with aryl halides
The heteroaryl derivatives (2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole) (2 mmol), the aryl halide derivatives (4-chloroacetophenone, 4-chloroanisole, 4-iodotoluene, 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene) (1 mmol), KOAc (1 mmol) and bis(NHC)PdX2 complexes 1a–h (0.003 mmol) were dissolved in N,N-dimethylacetamide (DMAc) (2 mL) in a small Schlenk tube under argon as described in the literature [9]. The reaction mixture was stirred in an oil bath at 130 °C for 1 h, and then was cooled to room temperature and the solvent was removed under vacuum. The obtained residue was purified by column chromatography (silica gel 60–120 mesh) using diethyl ether/n-hexane (1:5) as eluent to afford the pure product. The purity of the compounds was checked by gas chromatography (GC) and gas chromatography–mass spectrometry (GC–MS). Conversions were calculated by taking into account the conversion of aryl bromides to products.
Direct arylation of 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole with various aryl bromides
We performed some experiments for the parameters of direct arylation reaction of para-substituted aryl bromides with 2-n-butylfuran, 2-n-butylthiophene and 2-isopropylthiazole (for 1a–d) in the presence of 1a–h as catalyst. Conversions of the products for 2-n-butylfuran are between 59 and 99%, for 2-n-butylthiophene are between 74 and 98%, and for 2-isopropylthiazole are between 76 and 99% (Tables 2, 3, 4).
First, we examined the coupling of 2-n-butylfuran with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene, and 4-bromobenzene using complexes 1a–h as the catalyst. When the effect of 1a–h in the formation of the products 2, 3, 4 and 5 was analyzed, conversions of 86–99%, 59–96%, 75–93% and 73–96% were observed, respectively (Table 2). In the case of using 2-n-butylfuran with 4-bromoacetophenone and 4-bromobenzene, the desired coupling product is obtained in a better conversion, whereas when the 2-n-butylfuran was used with 4-bromoanisole and 4-bromotoluene, the desired coupling product is obtained in a lower conversion (Table 2). In general, the conversion at the reaction that used groups containing the electron donating substrates is lower, while the conversion at the reaction that used groups containing the electron withdrawing substrates is higher (Table 2).
Second, we examined the coupling of 2-n-butylthiophene with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene with complexes 1a–h as the catalyst. When the effect of 1a–h in the formation of product 6, 7, 8 and 9 was analyzed, conversions of 89–98%, 75–97%, 74–96% and 89–98% were observed, respectively (Table 3). The use of 4-bromoacetophenone with 2-n-butylthiophene gave the desired coupling product for different complexes (1c and 1d) as catalysts in better conversion than the others, such as 98 and 98%, respectively (Table 3). The use of 4-bromobenzene with 2-n-butylthiophene gave the desired coupling product for different complexes (1a and 1c) as catalysts in better excellent conversion than the others, such as 98% and 99%, respectively (Table 3). The 2-n-butylthiophene was bound with 4-bromoanisole to give the arylated products 7 in less conversions. When 4-bromoacetophenone, 4-bromotoluene and 4-bromobenzene with 2-n-butylthiophene were utilized in the direct arylation reaction, conversions of the products (6, 8 and 9) were obtained and observed to be better than the product 7 (Table 3).
Then, we investigated the coupling of 2-isopropylthiazole with 4-bromoacetophenone, 4-bromoanisole, 4-bromotoluene and 4-bromobenzene with complexes 1a–d as the catalyst. When the effect of 1a–d in the formation of product 10, 11, 12 and 13 was analyzed, conversions of 98–99%, 85–92%, 80–94% and 76–98% were observed, respectively (Table 4). The use of 4-bromoacetophenone with 2-isopropylthiazole gave the desired coupling product for all complexes 1a–d as catalysts in better excellent conversion than the others, such as 99%, 98%, 99%, and 99%, respectively (Table 4). When 4-bromoacetophenone, 4-bromotoluene and 4-bromobenzene with 2-isopropylthiazole were utilized in the direct arylation reaction, conversions of the products (10, 11 and 12) were obtained and observed to be better than the product 13 (Table 4). The 2-isopropylthiazole was bound with 4-bromoanisole to give the arylated products 13 in less conversions (Table 4).
Then, we used the chloride substrates, which are more difficult to bind than the bromide substrates. We examined the catalytic activities of 4-chloroacetophenone and 4-chloroanisole substrates in the presence 2-n-butylfuran and 2-n-butylthiophene with catalysts 1d and 1h at 16 h and 130 °C. As expected from the experiment, the conversions of coupling of 4-chloroacetophenone with 2-n-butylfuran and 2-n-butylthiophene (product 2 and 6) are higher than the conversions of coupling of 4-chloroanisol with 2-n-butylfuran and 2-n-butylthiophene (product 3 and 7) (Table 5). The use of catalyst 1h that containing aromatic substituent gave the desired coupling product in better conversion than the catalyst 1d that containing the aliphatic substituent. We can say reductive elimination may be easier for aromatic compounds. However, 4-iodotoluene, which easily bonded with 2-n-butylfuran and 2-n-butylthiophene (product 4 and 8) was obtained at low temperature (80 °C) and in a short time (30 min) in the high conversion (Table 6). All the above catalytic studies, when compared with the similar studies in the recent [31, 35, 39,40,41], the bis(NHC)PdX21a–h complexes appeared to be highly active catalysts in direct arylation reactions.
Conclusions
Conclusively, we reported the synthesis of the 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes 1a–h. The bis(NHC)PdX21a–h complexes were prepared from the Ag(I)NHC complexes via transmetallation method. The catalytic activity of this 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes have been investigated as they are more efficient and stable catalysts for the direct arylation reactions of 2-n-butylfuran 2-n-butylthiophene and 2-isopropylthiazole with aryl halide.
References
S. Diez Gonzalez (ed.), N-Heterocyclic Carbenes, From Laboratory Curiosities to Efficient Synthetic Tools (Royal Society of Chemistry, Cambridge, 2011)
M.N. Hopkinson, C. Richter, M. Schedler, F. Glorius, Nature. 510, 485–496 (2014)
C.S.J. Cazin (ed.), N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis (Springer, New York, 2011)
S.P. Nolan (ed.), N-Heterocyclic Carbenes in Effective Tools for Organometallic Synthesis (Wiley, Weinheim, 2014)
A.J. Arduengo, Acc. Chem. Res. 32, 913–921 (1999)
W. Liu, H. Cao, A. Lei, Angew. Chem. Int. Ed. 49, 2004–2008 (2010)
W.A. Herrmann, Angew. Chem. Int. Ed. 41, 1290–1309 (2002)
C.M. Crudden, D.P. Allen, Coord. Chem. Rev. 248, 2247–2273 (2004)
İ Özdemir, Y. Gök, Ö Özeroğlu, M. Kaloğlu, H. Doucet, Eur. J. Inorg. Chem. 12, 1798–1805 (2010)
D. Munz, C. Allolio, K. Döring, A. Poethig, T. Doert, H. Lang, T. Straßner, Inorg. Chim. Acta 392, 204–210 (2012)
C. Xu, X.-Q. Hao, Z. Li, X.-M. Dong, L.-M. Duan, Z.-Q. Wang, B.-M. Ji, M.-P. Song, Chem. Commun. 17, 34–37 (2012)
S. Demir, İ Özdemir, H. Arslan, D. VanDerveer, J. Organomet. Chem. 696, 2589–2593 (2011)
A.J. Arduengo, G. Bertrand, Chem. Rev. 109, 3209–3210 (2009)
D. Bourissou, O. Guerret, F.P.P. Gabbaï, G. Bertrand, Chem. Rev. 100, 39–92 (1999)
W.S. Hwang, I.J.B. Lin, Chem. Rev. 109, 3561–3598 (2009)
H. Arslan, İ Özdemir, D. Van Derveer, S. Demir, B. Çetinkaya, J. Coord. Chem. 62, 2591–2599 (2009)
Q. Ban, J. Zhang, T. Liang, C. Redshaw, W.-H. Sun, J. Organomet. Chem. 713, 151–156 (2012)
İ Özdemir, S. Demir, O. Şahin, O. Büyükgüngör, B. Çetinkaya, J. Organomet. Chem. 695, 1555–1560 (2010)
G. Dahm, C. Bailly, L. Karmazin, S. Bellemin-Laponnaz, J. Organomet. Chem. 794, 115–124 (2015)
A. Garoufis, S.K. Hadjikakou, N. Hadjiliadis, Coord. Chem. Rev. 253, 1384–1397 (2009)
A. Dervisi, Annu. Rep. Prog. Chem. Sect. A 106, 223–234 (2010)
A. Aktaş, S. Akkoç, Y. Gök, J. Coord. Chem. 66, 2901–2909 (2013)
S. Akkoç, İÖ İlhan, Y. Gök, V. Kayser, Inorg. Chim. Acta 461, 52–56 (2017)
S. Akkoç, Y. Gök, I. İlhan, V. Kayser, Beilstein J. Org. Chem. 12, 81–88 (2016)
C.B. Bheeter, L. Chen, J.-F. Soulé, H. Doucet, Catal. Sci. Technol. 6, 2005–2009 (2016)
S. Hayashi, T. Koizumi, Polym. Chem. 6, 5036–5039 (2015)
S. Hayashi, Y. Kojima, T. Koizumi, Polym. Chem. 6, 881–885 (2015)
T. Korenaga, R. Sasaki, K. Shimada, Dalton Trans. 44, 19642–19650 (2015)
D.S. Rampon, L.A. Wessjohann, P.H. Schneider, J. Org. Chem. 79, 5987–5992 (2014)
E. Iizuka, M. Wakioka, F. Ozawa, Macromolecules 49, 3310–3317 (2016)
S. Akkoç, Y. Gök, M. Akkurt, M.N. Tahir, Inorg. Chim. Acta 413, 221–230 (2014)
Y. Gök, S. Akkoç, S. Albayrak, M. Akkurt, M.N. Tahir, Appl. Organometal. Chem. 28, 244–251 (2014)
Z.-K. Xiao, H.-Y. Yin, J.-M. Lu, Inorg. Chim. Acta 423, 106–108 (2014)
S. Akkoç, Y. Gök, Inorg. Chim. Acta 429, 34–38 (2015)
M. Yiğit, B. Yiğit, Y. Gök, Inorg. Chim. Acta 453, 23–28 (2016)
M. Kaloğlu, İ Özdemir, V. Dorcet, C. Bruneau, H. Doucet, Eur. J. Inorg. Chem. 2017, 1382–1391 (2017)
S.-C. Yin, Q. Zhou, Q.-W. He, S.-W. Li, P.-C. Qian, L.-X. Shao, Tetrahedron 73, 427–431 (2017)
Q.-X. Liu, B.-Y. He, P.-C. Qian, L.-G. Shao, Org. Biomol. Chem. 15, 1151–1154 (2017)
M. Kaloğlu, N. Kaloğlu, İ. Özdemir, Chem. Select 3, 5600–5607 (2018)
M. Kaloğlu, İ Özdemir, Appl. Organometal. Chem. 32, e4399 (2018)
S. Akkoç, Y. Gök, Appl. Organometal. Chem. 28, 854–860 (2014)
Acknowledgements
This work was financially supported by Inönü University Research Fund (IUBAP 2011/21) and the authors acknowledge İnönü University Scientific and Technology Center for the elemental analyses of the compounds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gök, Y., Aktaş, A., Sarı, Y. et al. 2-methyl-1,4-benzodioxan-substituted bis(NHC)PdX2 complexes: Synthesis, characterization and the catalytic activity in the direct arylation reaction of some 2-alkyl-heterocyclic compounds. J IRAN CHEM SOC 16, 423–433 (2019). https://doi.org/10.1007/s13738-018-1512-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1512-y