
Abstract
Cylindromatosis (CYLD) is a tumor suppressor gene that is mutated in familial cylindromatosis, a rare autosomal dominant disorder associated with numerous benign skin adnexal tumors. CYLD is now known to regulate various signaling pathways, including transforming growth factor-β signaling, Wnt/β-catenin signaling, and NF-κB signaling by deubiquitinating upstream regulatory factors. Downregulation of CYLD has been reported in several malignancies; however, the clinical significance of CYLD expression in many malignancies, including breast cancer, remains to be elucidated. This study investigated the clinical significance of CYLD in breast cancer and its roles in tumor progression. We evaluated CYLD expression in matched normal breast tissue samples and tumor breast tissue samples from 26 patients with breast cancer and in a series of breast cancer cell lines. In addition, by means of immunohistochemistry, we investigated CYLD protein expression and its clinical significance in 244 breast cancer cases. We also analyzed the effects of CYLD repression or overexpression on breast cancer cell viability, cell migration, and NF-κB activity with or without receptor activator of NF-κB ligand (RANKL) stimulation. Breast cancer tissues demonstrated significantly reduced CYLD mRNA expression compared with normal breast tissues. Downregulation of CYLD promoted cell survival and migratory activities through NF-κB activation, whereas CYLD overexpression inhibited those activities in MDA-MB-231 cells. As an important finding, CYLD overexpression also inhibited RANKL-induced NF-κB activation. Our immunohistochemical analysis revealed that reduced CYLD protein expression was significantly correlated with estrogen receptor negativity, high Ki-67 index, high nuclear grade, decreased disease-free survival, and reduced breast cancer-specific survival in primary breast cancer. Moreover, reduced CYLD expression was an independent factor for poor prognosis in breast cancer. CYLD downregulation may promote breast cancer metastasis via NF-κB activation, including RANKL signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cylindromatosis gene (CYLD) was initially identified as a mutated gene in familial cylindromatosis, a rare autosomal dominant disorder associated with numerous benign skin adnexal tumors [1]. CYLD encodes a protein that possesses an ubiquitin C-terminal hydrolase domain, which allows the protein to function as a deubiquitinating enzyme [2]. CYLD is now known to regulate various signaling pathways, including transforming growth factor-β (TGF-β) signaling, Wnt/β-catenin signaling, and NF-κB signaling [3–5]. In the NF-κB signaling cascade, CYLD can negatively regulate various steps by deubiquitinating TNF receptor-associated factors 2 and 6, NF-κB essential modulator, receptor interacting protein 1, and TGF-β-activated kinase 1, which allows it to have important roles in inflammatory and immune reactions [2, 6–8]. Downregulation or mutation of CYLD has been reported in several malignancies [1, 9, 10], and Cyld knockout mice were shown to be highly susceptible to chemically induced skin tumors [11]. However, the clinical significance of CYLD expression in many malignancies, including breast cancer, remains to be elucidated.
Breast cancer, one of the most common human cancers, is a heterogeneous disease characterized by various molecular subtypes, which have distinct molecular features and clinical behaviors [12, 13]. Although some of these molecular features have been identified as biomarkers and drug targets, novel prognostic biomarkers and clarification of molecular mechanisms underlying the development and progression of breast cancer are still needed.
Constitutive activation of NF-κB is often observed in breast cancer tissues across the current major breast cancer subtypes (luminal, HER2, and triple negative) and cell lines with the more aggressive phenotypes [14–17]. Accumulating evidence has shown that activation of NF-κB promoted the development of breast cancer and resistance to conventional therapies such as endocrine therapy, cytotoxic chemotherapy, and radiotherapy [18–22]. Interestingly, recent studies showed that NF-κB signaling contributed to expansion of breast tumor stem cells and mammary tumorigenesis through cell-autonomous and non-cell-autonomous mechanisms [23–28]. In addition, receptor activator of NF-κB ligand (RANKL) and its receptor RANK, which activate the NF-κB pathway, have received much attention as key molecules in the promotion of breast cancer development and progression. Today, a drug targeting RANKL, denosumab, is registered to prevent skeletal-related events in advanced breast cancer patients with bone metastases in breast cancer treatment. However, no biomarker exists to identify tumors that are sensitive to the drug or to detect NF-κB activation, and the precise mechanisms underlying constitutive activation of NF-κB are not fully understood.
In this study, we investigated the clinical significance of CYLD expression in breast cancer and the roles of CYLD in breast cancer progression as related to NF-κB activation.
Materials and methods
Patients and tissues
Included in this study were paraffin-embedded breast cancer specimens from 244 women with histologically confirmed breast cancer (consecutive cases) who had received treatment at Kumamoto University Hospital between 2001 and 2008. Breast cancer oncologists chose the systemic treatments used, such as hormonal therapy, cytotoxic chemotherapy, and/or anti-HER2 therapy, according to the clinical evidence on tumor biology and clinical staging that was available at that time [29]. Our mRNA dataset of clinical breast cancer cases included 26 matched cases whose tumor tissues and adjacent normal breast tissues could both be evaluated. The ethics committee of Kumamoto University Graduate School of Medical Sciences approved the study protocol. Informed consent was obtained from all patients. In patients with primary invasive breast cancer (stages I to III, n = 230), 89.1 % of patients received any systemic adjuvant treatment (endocrine therapy 72.6 %; chemotherapy 37.4 %). Among them, patients with luminal/HER2-negative breast cancer (n = 162) received endocrine therapy (93.8 %) and/or chemotherapy (25.9 %). Regarding triple-negative cases (n = 32), 62.5 % of patients received chemotherapy, and 36.1 % of 36 patients with HER2-positive breast cancer received adjuvant trastuzumab.
Immunohistochemistry
Expression of CYLD in breast cancer samples was analyzed by using immunohistochemical methods with anti-CYLD antibody produced in rabbits (Sigma-Aldrich, St. Louis, MO). Expression of estrogen receptor (ER) α (SP1; Ventana Japan, Tokyo, Japan), progesterone receptor (PgR; 1E2; Ventana Japan), HER2 (Dako Japan, Tokyo, Japan), and Ki-67 (MIB-1; Dako Japan) was also examined according to the manufacturers’ recommended protocols. Experienced pathologists at our institution determined the nuclear grade according to nuclear pleomorphism and frequency of cell mitosis. The CYLD status was considered positive when cytoplasmic staining was ≥10 %. ER or PgR status was considered positive when nuclear staining was ≥1 % [30]. HER2 positivity was indicated by immunohistochemical staining score of 3+ (HercepTest, Dako) or by fluorescent in situ hybridization with a threshold ratio of more than 2.2 [31]. The Ki-67 index was scored according to the percentage of cells with nuclear staining of all cancer cells in the hot spots, at a ×400 high-power field, with at least 500 tumor cells counted.
Cell culture
Human breast adenocarcinoma cell lines (MDA-MB-231, MDA-MB-468, T47D, MCF7, ZR-75-30, MDA-MB-453, SK-BR-3, and HCC1569) and human mammary epithelial cells (HMEC) were purchased from American Type Culture Collection (Manassas, VA) and were passaged in our laboratory for less than 6 months after receipt or resuscitation. Cells were grown in Dulbecco’s modified Eagle medium (Gibco, Life Technologies Corporation, Carlsbad, CA) (MDA-MB-231and MDA-MB-453) or RPMI 1640 (MDA-MB-468, T47D, MCF7, ZR-75-30 and HCC1569) or McCoy’s 5a medium (SK-BR-3) supplemented with 10 % FBS (Gibco) under adherent conditions in 5 % CO2 at 37 °C. The details of these cell lines’ subtypes are as follows: basal-like subtype, MDA-MB-231 and MDA-MB-468; luminal subtype, MCF7 and T47D; luminal-HER2 subtype, ZR-75-30; and HER2 subtype, MDA-MB-453, SK-BR-3, and HCC1569.
RNA extraction and real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cultured cells by using the TRIzol Reagent (Life Technologies Corporation) and from tissue specimens by means of the RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacturers’ instructions. Total RNA was quantified via the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and was reverse transcribed to cDNA by using PrimeScript RT Master Mix (Takara Bio Inc., Shiga, Japan) according to the manufacturer’s protocol. All qRT-PCR reactions were performed with 2 μM cDNA and each primer at 0.2 μM via the LightCycler System (Roche Diagnostics, Tokyo, Japan) with SYBR Premix DimerEraser (Takara Bio Inc.).
Transfection with siRNA
Cell lines were transfected with CYLD-specific siRNA by using Lipofectamine 2000 (Life Technologies Corporation) according to the manufacturer’s protocol. Each analysis, such as total RNA extraction, protein extraction, and reporter assay, was performed after more than 48 h of incubation. Silencer Negative Control siRNA (Applied Biosystems, Life Technologies Corporation) was used as the control. CYLD-specific siRNA was sense 5′-GAUUGUUACUUCUAUCAAAtt-3′ and antisense 5′-UUUGAUAGAAGUAACAAUCtt-3′ (Applied Biosystems, Life Technologies Corporation).
Protein extraction and immunoblotting
Cells were washed once in ice-cold PBS (Gibco) and were then lysed by adding CelLytic M Cell Lysis Reagent (Sigma-Aldrich) containing a fresh Protease Inhibitor Cocktail (Sigma-Aldrich), 50 mM NAF, and 1 mM Na3VO4. After incubation for 15 min on a shaker on ice, cell lysate was removed from the dishes and was centrifuged at 15,000×g for 10 min to remove insoluble material. The protein concentration was determined by using the BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Rockford, IL) according to manufacturer’s instructions. Equal amounts of protein were fractionated via SDS-PAGE and fractions were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Primary antibodies used for immunoblotting were as follows: rabbit anti-CYLD antibody (Sigma-Aldrich), mouse anti-maspin antibody (Santa Cruz Biotechnology Inc., TX), and mouse anti-β-actin antibody (Sigma-Aldrich). Blots were visualized by using ECL Prime Western Blotting Detection Reagent (GE Healthcare Japan, Tokyo, Japan) according to the manufacturer’s instructions.
Reporter assay
Cell lines transfected with siRNA (negative control or CYLD) or plasmid (pcDNA or wild-type CYLD) were sequentially cotransfected with NF-κB reporter plasmid (pGL4b vector; Promega, Madison, WI) and control plasmid (phRG-TK vector; Promega). After a 24-h incubation in serum-free medium, cells were treated with recombinant human soluble RANKL (Peprotech, Rocky Hill, NJ) for 4 h or were untreated. Luciferase activities were determined with the Dual-Luciferase Reporter Assay System (Promega).
Cell viability, apoptosis, and migration assays
Cell viability was evaluated by means of the MTS assay using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) or Luna automated cell counter (Logos Biosystems, Annandale, VA). To evaluate apoptosis, we used the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Tokyo, Japan), according to the manufacturer’s protocol, and we performed the analysis with the FACSCalibur Flow Cytometer (BD Biosciences). We used the wound-healing assay to evaluate cell migration and quantified the results with ImageJ software (National Institutes of Health, Bethesda, MD).
Statistical analysis
We analyzed the data, presented as mean ± SD, by using Student’s t test or ANOVA. The means and SD were generated from triplicates in one experiment and then we confirmed the same results from two or three independent experiments. We utilized the nonparametric Wilcoxon, Fisher’s exact, and χ 2 tests for statistical analysis of the associations between CYLD status and clinicopathological factors. Disease-free survival (DFS) and breast cancer-specific survival (BCSS) curves were calculated according to the Kaplan–Meier method and were verified by means of the log-rank test. Univariate and multivariate analyses of prognostic values were performed with the Cox proportional hazards model. Statistical significance was defined as two-sided P < 0.05. JMP software version 8.0 for Windows (SAS Institute Japan, Tokyo, Japan) was used for all statistical analyses.
Results
CYLD expression in breast cancer
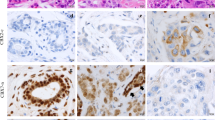
We first analyzed CYLD mRNA expression in matched tumor tissues and adjacent normal breast tissues from 26 patients with breast cancer. Breast cancer lesions had significantly lower expression of CYLD mRNA than did normal lesions [cancer: median 1.25 (25th–75th percentile 0.17–3.05); normal: median 9.60 (25th–75th percentile 7.08–11.82), paired Student’s t test, P < 0.0001] (Fig. 1a). Our immunohistochemical analysis of breast cancer tissues showed that CYLD expression occurred predominantly in normal appearing mammary epithelial cells, and reduced CYLD expression was found in cancer cells, as seen in a representative tissue section (Fig. 1b). All breast cancer cell lines also demonstrated significantly reduced CYLD mRNA expression, independent of molecular subtypes, compared with HMEC (Fig. 1c). These data indicate that CYLD mRNA expression was repressed in breast cancer.

CYLD expression in breast cancer. a CYLD mRNA expression in breast cancer tissue and the surrounding normal breast tissue of the same patients (n = 26). b Representative tissue section of breast cancer with CYLD downregulation. c CYLD mRNA expression in breast cancer cell lines and non-malignant HMEC. CYLD mRNA expression level was measured by means of qRT-PCR *P < 0.05 compared with HMEC in Student’s t test. Values are mean ± SD of triplicate samples
CYLD repression increased viability and migration of breast cancer cells
To determine the involvement of CYLD repression in breast cancer progression, we used siRNA to study the effects of CYLD knockdown on cell viability and motility. CYLD mRNA level was reduced by approximately 90 % 48 h after transfection of CYLD-specific siRNA in MDA-MB-231 cells (Fig. 2a). With MDA-MB-231, we observed increased cell viability (Fig. 2b), with decreased apoptosis in serum-free conditions (Fig. 2c), after transfection with CYLD-specific siRNA. No significant change in the cell cycle occurred (Fig. 2d). In addition, CYLD knockdown in MDA-MB-231 cells stimulated migration (Fig. 2e). The same results were also obtained in MDA-MB-468 cells (data not shown). In contrast, overexpression of wild-type CYLD resulted in decreased cell viability, increased apoptosis in serum-free condition and reduced migration (Fig. 2f–h). Furthermore, we observed enhanced expression of various genes encoding TNFα, interleukin-8, and granulocyte–macrophage colony-stimulating factor, which are regulated by NF-κB and associated with breast cancer progression, after CYLD knockdown (Fig. 2i–K). We also found that downregulation of CYLD led to reduced expression of maspin, which was reportedly a crucial suppressor of breast cancer metastasis (Fig. 2l) [32, 33].

Effects of CYLD expression on cell survival and migration in MDA-MB-231 cells. a Knockdown efficiency of CYLD mRNA by siRNA in MDA-MB-231 cells. b Cell viability in serum-free medium was evaluated by MTS assay 72 h after transfection with siRNA. c Anti-apoptotic property in serum-free condition was evaluated by Annexin V assay 96 h after transfection with siRNA. d Cell cycle distribution in serum-starved condition 72 h after siRNA transfection was analyzed by flow cytometry following staining with propidium iodide. e Cells were wounded at 72 h after siRNA transfection. After incubation for 48 h in serum-free medium, cell migration was analyzed. Effects of CYLD overexpression on cell viability (f) and apoptosis (g) were assessed by MTS assay and Annexin V assay, respectively, 96 h after transfection with wild-type CYLD plasmid. h Cells were wounded at 96 h after transfection with wild-type CYLD plasmid. After incubation for 24 h in serum-free medium, cell migration was measured. i, j, and k Expression level of TNFa (i), IL-8 (j), or GM-CSF (k) mRNA was measured by means of qRT-PCR 72 h after siRNA transfection. *P < 0.05 in Student’s t test. Values are mean ± SD of triplicate samples. l Maspin protein expression was evaluated via western blotting 72 h after siRNA transfection
Negative regulation by CYLD of NF-κB activity including RANKL-RANK signaling in breast cancer
To determine whether CYLD expression was associated with NF-κB activity in breast cancer, we utilized the reporter assay. CYLD knockdown in MDA-MB-231, MDA-MB-468, and T47D cells stimulated NF-κB activity (Fig. 3a and Supplementary Fig. S1), whereas overexpression of wild-type CYLD suppressed it (Fig. 3b). In line with the above finding, the increased cell viability and migratory activity induced by CYLD knockdown were abolished by adding MG132 as an inhibitor of NF-κB signaling (Fig. 3c, d).

CYLD as a negative regulator of NF-κB signaling including RANKL in MDA-MB-231 cells. a and b NF-κB activity was evaluated by luciferase reporter assay 96 h after transfection with siRNA or vectors in serum-free condition. *P < 0.05 in Student’s t test. c NF-κB inhibitor, MG132 (0.2 μM), was added to the cells at 48 h after siRNA transfection, followed by incubation for up to 48 h in serum-free condition. Cell viability was evaluated by MTS assay. d Cells were wounded at 72 h after siRNA transfection. After incubation with 1 μM MG132 for 24 h, cell migration was analyzed. *P < 0.05 compared with control siRNA treated with DMSO, and **P < 0.05 compared with CYLD siRNA treated with DMSO in Student’s t test. e Recombinant RANKL (500 ng/ml) was added to the cells 96 h after siRNA transfection. After incubation for 4 h in serum-free condition, NF-κB activity was evaluated by luciferase reporter assay. f Cells were wounded at 96 h after siRNA transfection. After incubation with recombinant RANKL (500 ng/ml) for 24 h, cell migration was analyzed. *P < 0.05 compared with control siRNA and mock treatment, and **P < 0.05 compared with CYLD siRNA and mock treatment in Student’s t test. g Recombinant RANKL (500 ng/ml) was added to the cells 96 h after transfection with wild-type CYLD plasmid. After incubation for 4 h in serum-free condition, NF-κB activity was evaluated by luciferase reporter assay. *P < 0.05 compared with all the other conditions in Student’s t test
Recent studies also showed that RANKL-NF-κB signaling had crucial roles in the development and progression of breast cancer other than bone events [34–38]. Thus, we next investigated whether CYLD was involved in RANKL-induced NF-κB activation in MDA-MB-231 cells. We found that RANKL enhanced not only NF-κB activity (Fig. 3e) but also migration of both control and CYLD knockdown cells (Fig. 3f). An important finding was that RANKL-induced NF-κB activation was inhibited by overexpression of wild-type CYLD (Fig. 3g). These data indicate that CYLD was a negative regulator of NF-κB activation, including RANKL signaling, in breast cancer cells.
CYLD was an independent prognostic factor in primary breast cancer
We also investigated correlations between CYLD protein expression and clinicopathological factors and prognosis in breast cancer. Our immunohistochemical analysis (n = 244) indicated CYLD-negative results in 73 cases (29.9 %) and CYLD-positive results in 171 cases (70.1 %) (Fig. 4a, b). CYLD-negative status occurred more frequently in ER-negative (Fisher’s exact test, P = 0.024), PgR-negative (Fisher’s exact test, P = 0.008), or triple-negative (χ 2 test, P = 0.032) breast cancer tissues [12] than did CYLD-positive status (Table 1). CYLD-negative status also correlated with a high Ki-67 index (χ 2 test, P = 0.009) and a high nuclear grade (χ 2 test, P < 0.001).

Relationship between CYLD expression and prognosis in primary invasive breast cancer. CYLD protein expression in primary invasive breast cancer tissues (n = 244) was analyzed by immunohistochemistry. a and b Representative examples of CYLD-positive breast cancer tissue (a) and CYLD-negative tissue (b). Note that normal mammary epithelial cells or breast cancer cells mainly expressed CYLD. c and d Kaplan–Meier plots for disease-free survival (DFS) (c) and breast cancer-specific survival (BCSS) (d) according to CYLD status (negative, N = 70; positive, N = 160) were constructed in invasive primary breast cancer cases (n = 230) except in situ and metastatic cases. Statistical significances were evaluated using log-rank test
Kaplan–Meier survival analysis (n = 230) revealed that, with a median follow-up of 65.7 months, reduced CYLD expression correlated strongly with poor DFS (log-rank test, P = 0.0005, Fig. 4c) and poor BCSS (log-rank test, P = 0.0009, Fig. 4d) in patients with primary invasive breast cancer (stages I–III). The 5-year DFS and BCSS rates were 74.9 and 82.6 % in patients with CYLD-negative tumors compared with 91.0 and 95.3 % in patients with CYLD-positive tumors, respectively. Similar results were obtained for each breast cancer subtype (luminal A, luminal B, and triple negative) (Supplementary Fig. S2), except HER2 subtype [12]. With regard to CYLD expression and DFS, we obtained the same results when we evaluated CYLD mRNA expression (data not shown). Furthermore, our multivariate analysis using the Cox proportional hazards model revealed that CYLD expression was an independent prognostic factor for DFS (CYLD-negative status, hazard ratio HR 2.43; 95 % confidence interval CI 1.16–5.26, P = 0.019) (Table 2). Multivariate analysis also showed a tendency for a strong relationship between CYLD expression and BCSS (Cox proportional hazards model, P = 0.057, Supplementary Table S1).
Discussion
In this study, we demonstrated for the first time that reduced CYLD expression was an independent factor for poor prognosis in breast cancer. Loss of CYLD stimulated breast cancer cell viability and migration by means of NF-κB activation. In addition, CYLD negatively regulated RANKL-induced NF-κB activation and migration. Our findings thus suggest that CYLD downregulation promotes breast cancer metastasis via activation of the NF-κB pathway including RANKL signaling.
Despite recent data showing that CYLD regulated diverse signaling pathways including the NF-κB pathway, the clinical significance of CYLD expression in malignancies has remained largely unknown. This report is the first to provide analyses of CYLD expression in breast cancer, with reduced CYLD expression being associated with poor prognosis. These findings are consistent with data from a public database of gene expression arrays (Supplementary Fig. S3A) [39]. Our data and the database finding that CYLD had a more significant prognostic value in patients who had had systemic treatment of any kind compared with those who had not had such treatment (Supplementary Fig. S3B and C). Further investigation focused on association between CYLD and drug resistance for chemotherapy or endocrine therapy may provide beneficial information about breast cancer treatment.
Reduced CYLD expression was significantly associated with more aggressive clinicopathological parameters such as ER-negative status, high Ki-67 index, and high nuclear grade, which indicates the tumor-suppressive effects of CYLD. Our multivariate analysis demonstrated that reduced CYLD expression was an independent prognostic factor for DFS. These findings suggest that downregulation of CYLD commonly has tumor-promoting effects in breast cancer, in a manner similar to that of activated NF-κB [14–17]. Previous clinical studies showed that NF-κB activation in breast cancer occurred more frequently in ER-negative cases or high-risk ER-positive cases, although the mechanisms of the NF-κB activation remain unknown [15, 16]. Additional studies to evaluate NF-κB activity in breast cancer tissues in relation to CYLD expression may clarify the relevance of the NF-κB pathway in breast cancer progression. Although the mechanisms of CYLD downregulation remain to be elucidated, our clinical findings suggest that CYLD is a useful prognostic factor and a potent tumor suppressor in breast cancer.
Our clinical results are supported by in vitro data showing that downregulation of CYLD expression led to acquisition of more aggressive features of breast cancer, including increased survival and migration of breast cancer cells. Consistent with previous studies [2, 7, 8, 11, 40], our analysis indicated that CYLD negatively regulated NF-κB activity. Our data thus suggest that CYLD downregulation promotes metastasis and/or recurrence of breast cancer through induction of NF-κB activation.
Various cytokines and growth factors activate NF-κB [18]. Certain studies showed that RANKL-RANK signaling had crucial roles in development and progression of breast cancer. The RANKL-RANK system was originally shown to be essential for osteoclastogenesis and physiological bone remodeling and to promote bone metastasis in malignancies including breast cancer [41, 42]. Recently, in advanced breast cancer, anti-RANKL antibody, denosumab, is approved in prevention of skeletal-related events in patients with bone metastases from solid tumors. However, more recent studies revealed that RANKL signaling in breast epithelial cells themselves promoted breast carcinogenesis and metastasis to sites other than bone in vivo [35–37] and induced stemness and epithelial-mesenchymal transition [34–38], which suggests broader than expected effects of denosumab on breast cancer progression [43]. Biomarkers that predict a dependence on RANKL signaling should hold promise for treatment, but the clinical significance of RANKL-RANK expression in breast cancer is still controversial [44, 45]. As an important result in our study, CYLD inhibited RANKL-induced NF-κB activation even in breast cancer cells, a finding that is consistent with another result reported for osteoclasts [46]. In addition, downregulation of CYLD enhanced RANKL-induced migration of breast cancer cells. Additional studies to clarify the exact roles of CYLD in breast cancer, especially in RANKL-induced malignant phenotypes, may contribute to our understanding of the molecular mechanisms underlying this disease, including dysregulated NF-κB activation, and the development of improved anti-RANKL therapy.
In conclusion, reduced expression of CYLD was an independent factor for poor prognosis in breast cancer. CYLD downregulation may contribute to breast cancer metastasis through activation of NF-κB including RANKL signaling. Elucidation of CYLD functions in malignant tumors is therefore warranted.
Abbreviations
- CYLD:
-
Cylindromatosis
- RANKL:
-
NF-κB ligand
- HER2:
-
Human epidermal growth factor receptor 2
- ER:
-
Estrogen receptor
- PgR:
-
Progesterone receptor
- HMEC:
-
Human mammary epithelial cells
References
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S (2000) Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 25(2):160–165. doi:10.1038/76006
Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G (2003) The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424(6950):801–805. doi:10.1038/nature01802
Wang WY, Lim JH, Li JD (2012) Synergistic and feedback signaling mechanisms in the regulation of inflammation in respiratory infections. Cell Mol Immunol 9(2):131–135
Lim JH, Jono H, Komatsu K, Woo CH, Lee J, Miyata M, Matsuno T, Xu X, Huang Y, Zhang W, Park SH, Kim YI, Choi YD, Shen H, Heo KS, Xu H, Bourne P, Koga T, Yan C, Wang B, Chen LF, Feng XH, Li JD (2012) CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat Commun 3:771
Tauriello DV, Haegebarth A, Kuper I, Edelmann MJ, Henraat M, Canninga-van Dijk MR, Kessler BM, Clevers H, Maurice MM (2010) Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell 37(5):607–619
Harhaj EW, Dixit VM (2012) Regulation of NF-kappaB by deubiquitinases. Immunol Rev 246(1):107–124. doi:10.1111/j.1600-065X.2012.01100.x
Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G (2003) CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424(6950):793–796. doi:10.1038/nature01803
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R (2003) Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424(6950):797–801. doi:10.1038/nature01811
Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL (2007) Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 12(2):131–144
Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK (2007) Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 28(1):21–27
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R (2006) Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 125(4):665–677
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ (2011) Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann Oncol 22(8):1736–1747
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874. doi:10.1073/pnas.191367098
Yamaguchi N, Ito T, Azuma S, Ito E, Honma R, Yanagisawa Y, Nishikawa A, Kawamura M, Imai J, Watanabe S, Semba K, Inoue J (2009) Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci 100(9):1668–1674
Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, Benz CC (2005) Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int J Biochem Cell Biol 37(5):1130–1144
Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD (2004) NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A 101(27):10137–10142. doi:10.1073/pnas.0403621101
Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE (1997) Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest 100(12):2952–2960. doi:10.1172/JCI119848
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441(7092):431–436
Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T (2004) NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 114(4):569–581. doi:10.1172/JCI21358
deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, Freeman JW, Weiss GR (2004) NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol 15(6):885–890
Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I (2002) p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell 1(5):493–503
Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr (1999) Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med 5(4):412–417. doi:10.1038/7410
Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J (2013) NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun 4:2299
Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS (2013) Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene
Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A, Hall CL, Palanisamy N, Heath AN, Tawakkol N, Luther TK, Clouthier SG, Chadwick WA, Day ML, Kleer CG, Thomas DG, Hayes DF, Korkaya H, Wicha MS (2013) HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer Res 73(5):1635–1646
Hinohara K, Kobayashi S, Kanauchi H, Shimizu S, Nishioka K, Tsuji E, Tada K, Umezawa K, Mori M, Ogawa T, Inoue J, Tojo A, Gotoh N (2012) ErbB receptor tyrosine kinase/NF-kappaB signaling controls mammosphere formation in human breast cancer. Proc Natl Acad Sci USA 109(17):6584–6589
Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, Ojeifo J, Jiao X, Yeow WS, Katiyar S, Shirley LA, Joyce D, Lisanti MP, Albanese C, Pestell RG (2010) The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res 70(24):10464–10473
Kai K, Arima Y, Kamiya T, Saya H (2010) Breast cancer stem cells. Breast Cancer 17(2):80–85. doi:10.1007/s12282-009-0176-y
Goldhirsch A, Wood WC, Gelber RD, Coates AS, Thurlimann B, Senn HJ (2007) Progress and promise: highlights of the international expert consensus on the primary therapy of early breast cancer 2007. Ann Oncol 18(7):1133–1144
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, Wittliff JL, Wolff AC (2010) American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 28(16):2784–2795
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF (2007) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 25(1):118–145
Affara NI, Coussens LM (2007) IKKalpha at the crossroads of inflammation and metastasis. Cell 129(1):25–26
Maass N, Hojo T, Rosel F, Ikeda T, Jonat W, Nagasaki K (2001) Down regulation of the tumor suppressor gene maspin in breast carcinoma is associated with a higher risk of distant metastasis. Clin Biochem 34(4):303–307
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A, Climent F, Soler MT, Munoz P, Vinals F, Tometsko M, Branstetter D, Dougall WC, Gonzalez-Suarez E (2012) RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res 72(11):2879–2888
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470(7335):548–553
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, Pasparakis M, Khokha R, Ormandy CJ, Widschwendter M, Schett G, Penninger JM (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468(7320):98–102
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, Pinkas J, Branstetter D, Dougall WC (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468(7320):103–107
Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, Visvader JE (2010) Control of mammary stem cell function by steroid hormone signalling. Nature 465(7299):798–802
Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123(3):725–731. doi:10.1007/s10549-009-0674-9
Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, Mosialos G, Li JD (2004) NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem 279(35):36171–36174. doi:10.1074/jbc.M406638200
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, Komnenovic V, Kong YY, Schreiber M, Dixon SJ, Sims SM, Khokha R, Wada T, Penninger JM (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440(7084):692–696
Ohshiba T, Miyaura C, Inada M, Ito A (2003) Role of RANKL-induced osteoclast formation and MMP-dependent matrix degradation in bone destruction by breast cancer metastasis. Br J Cancer 88(8):1318–1326. doi:10.1038/sj.bjc.6600858
Brown JE, Coleman RE (2012) Denosumab in patients with cancer-a surgical strike against the osteoclast. Nat Rev Clin Oncol 9(2):110–118
Cross SS, Harrison RF, Balasubramanian SP, Lippitt JM, Evans CA, Reed MW, Holen I (2006) Expression of receptor activator of nuclear factor kappabeta ligand (RANKL) and tumour necrosis factor related, apoptosis inducing ligand (TRAIL) in breast cancer, and their relations with osteoprotegerin, oestrogen receptor, and clinicopathological variables. J Clin Pathol 59(7):716–720
Bhatia P, Sanders MM, Hansen MF (2005) Expression of receptor activator of nuclear factor-kappaB is inversely correlated with metastatic phenotype in breast carcinoma. Clin Cancer Res 11(1):162–165
Jin W, Chang M, Paul EM, Babu G, Lee AJ, Reiley W, Wright A, Zhang M, You J, Sun SC (2008) Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest 118(5):1858–1866. doi:10.1172/JCI34257
Acknowledgments
The authors thank Dr. Seiji Okada and Dr. Manabu Taura for supplying pGL4b and phRG-TK vectors; Y. Ogasawara, K. Kai, M. Imamoto, E. Nakamura, and H. Katsura for excellent technical support; Dr. K. Iyama, Dr. Y. Honda, and Dr. T. Asato for their excellent pathological diagnosis. The authors’ work was supported by a Grand-in-Aid for Scientific Research (A) 24249036 (Y. Ando), by Grand-in-Aid for Young Scientists (B) 23790091 (H. Jono) and (B) 24792238 (S. Shinriki) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of interest
The authors disclosed no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hayashi, M., Jono, H., Shinriki, S. et al. Clinical significance of CYLD downregulation in breast cancer. Breast Cancer Res Treat 143, 447–457 (2014). https://doi.org/10.1007/s10549-013-2824-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2824-3