
Abstract
Maple syrup urine disease (MSUD) is an autosomal recessive inherited disorder that affects branched-chain amino acid (BCAA) catabolism and is associated with acute and chronic brain dysfunction. Recent studies have shown that inflammation may be involved in the neuropathology of MSUD. However, these studies have mainly focused on single or small subsets of proteins or molecules. Here we performed a case-control study, including 12 treated-MSUD patients, in order to investigate the plasmatic biomarkers of inflammation, to help to establish a possible relationship between these biomarkers and the disease. Our results showed that MSUD patients in treatment with restricted protein diets have high levels of pro-inflammatory cytokines [IFN-γ, TNF-α, IL-1β and IL-6] and cell adhesion molecules [sICAM-1 and sVCAM-1] compared to the control group. However, no significant alterations were found in the levels of IL-2, IL-4, IL-5, IL-7, IL-8, and IL-10 between healthy controls and MSUD patients. Moreover, we found a positive correlation between number of metabolic crisis and IL-1β levels and sICAM-1 in MSUD patients. In conclusion, our findings in plasma of patients with MSUD suggest that inflammation may play an important role in the pathogenesis of MSUD, although this process is not directly associated with BCAA blood levels. Overall, data reported here are consistent with the working hypothesis that inflammation may be involved in the pathophysiological mechanism underlying the brain damage observed in MSUD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maple syrup urine disease (MSUD) is an autosomal recessive amino acid disorder caused by mutations in genes that encode the components of the branched-chain α-ketoacid dehydrogenase (BCKDH) complex, which catalyzes the first irreversible step in branched-chain amino acid (BCAA) catabolism (Menkes 1959; Dancis et al 1977). The resulting metabolic block leads to elevated plasma concentrations of BCAA (leucine, isoleucine and valine) and the corresponding branched-chain α-ketoacids (α-ketoisocaproic acid, α-keto-β-methylvaleric acid and α-ketoisovaleric acid) (Treacy et al 1992; Chuang and Shih 2001). If untreated, affected individuals accumulate substantial BCAA and their corresponding ketoacids—mostly leucine and α-ketoisocaproic acid—and suffer encephalopathy within the first week of life as well as cerebral oedema and dysmyelination with chronic brain injury (Crome et al 1961; Silberman et al 1961; Chuang and Shih 2001). The global frequency is approximately one in 185,000 live newborns (Mackenzie and Woolf 1959; Chuang and Shih 2001; Zinnanti et al 2009), although data retrieved from newborn screenings suggest this rate can be higher (Simon et al 2006; Fingerhut 2009; Quental et al 2010).
Although numerous factors and mechanisms have been proposed to understand the pathogenesis of MSUD, its aetiopathology remains unknown. However, several studies have proposed that leucine and/or its ketoacid are the main neurotoxic metabolites in MSUD since the appearance of neurological symptoms is related to increased plasma concentrations of these compounds. Indeed, the elevated BCAA level may saturate the LAT-1 transporter and block the uptake of other large neutral amino acids, leading to decreased synthesis of neurotransmitters such as dopamine, serotonin, norepinephrine, and histamine (Wajner and Vargas 1999; Tavares et al 2000; Wajner et al 2000; Araujo et al 2001; Zielke et al 2002). Moreover, it has been demonstrated that metabolite accumulation in MSUD causes mitochondrial bioenergetics dysfunction (Sgaravatti et al 2003; Funchal et al 2006a, b; Wajner et al 2007; Ribeiro et al 2008; Amaral et al 2010), oxidative stress (Bridi et al 2003, 2005; Funchal et al 2006a, b; Barschak et al 2008, 2009; Mescka et al 2013, 2015a, b; Sitta et al 2014), apoptosis of neural cells (Jouvet et al 1998, 2000a, b), and alterations in the cholinergic system (Scaini et al 2012), in neurotrophin levels (Scaini et al 2013a, b, 2015) and in lysosomal proteases (Scaini et al 2016).
Inflammation has been recognized as a risk factor for age-related neurodegenerative diseases (Amor et al 2010, 2014; Fakhoury 2015), and high inflammatory profiles at baseline assessment may increase risk of conversion to dementia at long-term follow-up (Schmidt et al 2002; Cunningham and Hennessy 2015). A growing body of evidence demonstrates that higher levels of circulating inflammatory markers, especially interleukin-6 (IL-6) and C-reactive protein, are associated with greater cognitive decline (Yaffe et al 2003; Singh-Manoux et al 2014; Palta et al 2015). Moreover, studies have shown that modulators of the peripheral immune system can induce psychiatric symptoms in animal models and humans (Dantzer et al 2008; Laske et al 2008; Harrison et al 2009; Eisenberger et al 2010; Raison and Miller 2011). For example, healthy participants exposed to low-dose endotoxin (Escherichia coli) showed greater increases in self-reported and observer-rated depressed mood, suggesting that inflammation alters reward-related neural responses in humans and these in turn mediate the effects of inflammation on depressed mood (Eisenberger et al 2010). In this line, previous studies have shown that inflammatory processes play an integral role in the pathophysiology of certain inborn errors of metabolism, including phenylketonuria (Deon et al 2015), glutaric acidemia type I (Seminotti et al 2016), and methylmalonic acidemia (Ribeiro et al 2013). It has been shown that patients with MSUD treated with restricted protein diets have high levels of pro-inflammatory cytokines (Mescka et al 2015a, b). Moreover, studies using animal models and cell cultures have also demonstrated that BCAAs alter the balance between pro-inflammatory and anti-inflammatory cytokines (De Simone et al 2013; Rosa et al 2016).
Considering the hypothesis that BCAAs themselves and/or one of their metabolites could be responsible for MSUD’s central nervous system (CNS) dysfunction and that there are few studies about inflammation in this disease, the aim of this study was to investigate plasma biomarkers of inflammation from patients with MSUD in treatment to establish a possible relationship between these biomarkers and the disease.
Material and methods
Sample and subjects
Plasma specimens from 12 treated patients with MSUD with the classic form and nine healthy individuals (control group) were used to evaluate the inflammatory markers and concentrations of amino acids. The diagnosis of MSUD was based on the high concentration of leucine and alloisoleucine in plasma, or by DNA analysis. The patients were between 15 days and 2 months old at diagnosis and followed a treatment that consisted of a natural, protein-restricted diet with low BCAAs and supplemented with a semi-synthetic formula of essential amino acids containing small amounts of vitamins and minerals. In addition, the patients with MSUD were supplemented with isoleucine and valine capsules (Table 1). Table 2 displays the age at diagnosis, age at testing, length of treatment, genotypes, and clinical profiles of the patients with MSUD at presentation and under treatment. In addition, this study included a control group, comprising nine subjects, age- and gender-controlled. The present study was approved by the Ethics Committee of the Hospital de Clínicas de Porto Alegre, RS, Brazil. All parents of the patients included in the present study provided informed consent according to the guidelines of our committee.
Plasma preparation
Human blood samples were collected in heparin-coated collection tubes from fasting patients with MSUD (2 h) or nine healthy individuals (control group) by venous puncture during the baseline visits. Whole blood was centrifuged at 1000×g, and plasma was removed by aspiration and frozen at −80 °C until analysis.
Amino acid determination
The free amino acids in plasma were determined by HPLC according to Joseph and Marsden (1986), with slight modifications (Wajner et al 2000). Amino acids were quantified by comparing their chromatographic peak area to those obtained from a known standard mixture and to that of an internal standard peak area (homocysteic acid).
Gene sequencing
A DNA sample was extracted from the peripheral blood of each proband and their biological parents. BCKDHA, BCKDHB, and DBT were amplified by polymerase chain reaction (PCR) with specific oligonucleotides, including exons and exon-intron boundaries. The amplification products were purified and sequenced by Sanger method in an ABI 3130 Genetic Analyzer (Applied Biosystems). Genetic variants were identified by comparison with reference sequences NG_013004.1 (BCKDHA), NG_009775.1 (BCKDHB), and NG_011852.2 (DBT) (GenBank/NCBI). Most pathogenic variations occur within coding regions or consensus splice and branch sites, and thus our approach can detect most but not all mutations. Moreover, this method does not detect large genomic deletions or deletions involving oligonucleotide primer sequences.
Inflammatory markers
Samples were randomized based on diagnostic group and plasma inflammatory markers (GM-CSF, RANTES, sICAM1, sVCAM1, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, INF-ɣ, and TNF-α) were assayed in duplicate with Luminex xMAP Technology (Millipore, USA & Canada). The xMAP platform used here was based on the Rules-Based Medicine (RBM) fluorescent beads and antibody pairs. These are sensitive, specific, and widely used reagents sourced by numerous manufacturers, and data collected using xMAP multiplex beads have been widely reported in the literature in studies with simultaneous assay of multiple proteins.
The assays were conducted in 96-well polystyrene, round-bottom microplates. Initially, 200 μL of wash buffer was added to each well of the plate, and the plate was sealed and mixed on a plate shaker for 10 min at room temperature. Subsequently, 50 μL of the standard and 25 μL of the sample were added to the appropriate wells. A 25-μL aliquot of the working bead mixture was transferred into the wells. The plate was incubated with agitation on a plate shaker overnight (16–18 h) at 4 °C. The plate was then placed into the magnetic separator and left to separate for 60 s. The supernatant was carefully removed from each well by manual inversion. Beads were washed three times by adding 100 μL of assay buffer into each well to ensure absence of any undesirable or non-specifically bound antibodies. Following the protocol, 50 μL of a detection antibody was added to each well. Incubation was again performed in darkness and at RT on a plate shaker (850 rpm) for 60 min. Finally, 50 μL of streptavidin-PE was added to each well. The plate was incubated on a plate shaker (850 rpm) in the dark at RT for 30 min. The supernatant was carefully removed after magnetic separation of the beads by manual inversion, and washing was performed as previously described. Sheath fluid (150 μL) was added into each well, and the plate was placed onto a plate shaker for approximately 5 min to achieve gentle agitation of the beads. Samples were run in duplicate using a Luminex 200™ system (Millipore, USA & Canada), and data analysis was conducted in MILLIPLEX® Analyst 5.1 software using a 5-parameter logistic regression model. Inflammatory markers were expressed as pg/mL.
Statistical analysis
The association between dichotomous variables was assessed with the Fisher exact test. All variables were tested for Gaussian distribution by the Kolmogorov-Smirnov normality test. The initial comparisons between the two groups (patients vs. controls) were determined using Mann-Whitney or Student’s t tests when non-normally or normally distributed, respectively. Linear regression was used to adjust for possible confounding variables. We considered all variables associated with diagnostic group and biomarkers with p < 0.20 as possible confounding factors (Victora et al 1997). Spearman’s Rho(r) correlation analyses were performed to examine the relationship between clinical variables and plasma levels of inflammatory markers. Data are presented as the means and 95% confidence intervals. All statistical tests were two-tailed and were performed using a significance level of α = 0.05. Statistical analyses were performed using SPSS software version 22.0 (SPSS Inc., Chicago, IL, USA) as well as GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
The variants involving premature stop codons and deletions as observed in patients 3, 8, and 10 usually cause a substantial impact on the enzyme activity, with significant or total loss of function. These findings are consistent with the phenotype observed in “age of diagnosis” of the patients, with severe symptoms (Table 2). However, the p.Pro200Ter mutation in the BCKDHB gene has already been associated with a mild phenotype (Henneke et al 2003). The p.Lys166Asn and p.Arg168Cys mutations in the BCKDHB gene have already been associated with the classical phenotype of the disease (Imtiaz et al 2017). Functional or expression studies of alleles have not been performed. Maintaining good metabolic control and early diagnosis does not provide knowledge on the natural history of the disease and the phenotypic impact of the mutations found. Flaschker et al (2007) suggest that changes in the BCKDHA gene are more severe than in the BCKDHB and DTB genes; however, this observation is directly related to the mutations found, and many patients with alterations in the last two genes present the severe phenotype. The compound heterozygotes, (as observed in many of our patients) present the combination of the two different altered alleles, affecting individual predictions about alleles.
Notably, no significant differences in age or gender emerged between groups (Table 3). The levels of leucine in the MSUD group were 166.43 μmol/l ± 27.85, and their mean isoleucine and valine levels were 180.00 μmol/l ± 27.72 and 207.08 μmol/l ± 48.92, respectively. At the time of the study, all patients with MSUD were in treatment with a protein-restricted diet with isoleucine and valine supplementation as shown in Table 1. In this context, no significant alterations in leucine and valine levels were found between the healthy controls and the patients with MSUD, while isoleucine levels were significantly higher in the patients with MSUD. Moreover, phenylalanine, serine, ornithine, glutamate, and aspartate levels were significantly reduced, and glycine/threonine/arginine levels were increased in the patients with MSUD compared with the healthy controls (Table 3).
The role of inflammatory markers in MSUD was investigated by analyzing the protein levels of GM-CSF, RANTES, sICAM1, sVCAM1, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, INF-ɣ, and TNF-α using a Luminex xMAP kit. As seen in Table 4, a significant increase in pro-inflammatory cytokines IL-1β and IL-6 were found in the patients with MSUD compared to the control group in the adjusted analyses. However, no significant alterations in the levels of IL-2, IL-4, IL-5, IL-7, IL-8, and IL-10 were found between the healthy controls and the patients with MSUD. On the other hand, we found increased levels of TNF-α and INF-ɣ as well as an increase in cell adhesion molecules, sICAM1 and sVCAM1 in the patients with MSUD when compared to the controls. In addition, IL-1β, IL-6, TNF-α, INF-ɣ, sICAM1, and sVCAM1 remained significantly increased in the patients with MUSD in comparison with the control group after controlling for age (linear regression model). To further investigate the cytokine profile in MSUD, we also compared cytokine ratios. The patients with MSUD showed higher IFN-γ/IL-4, IL-2/IL-4, and IFN-γ/IL-10 ratios than the controls, suggesting a bias toward a Th1 profile. There was no significant difference regarding the IL-6/IL-10 ratio and TNF/IL-10 ratio.
Finally, to explore the potential clinical correlates of our findings, we tested possible correlations between the number of metabolic crises and LNAA levels with inflammatory markers. Our results showed a positive correlation between the number of metabolic crises and IL-1β levels (rho = 0.788, p = 0.0052, Fig. 1a) and sICAM-1 (rho = 0.816, p = 0.0030, Fig. 1b). However, no associations were found between protein levels of IL-6, TNF-α, INF-ɣ, and sVCAM1 and metabolic crisis (Suppl. Table 1). Moreover, the results of Spearman’s rank correlation showed no correlations between LNAA blood levels and IL-1β, IL-6, TNF-α, INF-ɣ, sICAM1, and sVCAM1 levels (Suppl. Table 2).

Correlation coefficient of IL-1β (a) and sICAM-1 (b) with the number of metabolic crises in controls and treated patients with MSUDs. The results were assessed using Spearman’s Rho(r) correlation analyses
Discussion
MSUD treatment involves long-term dietary management by restricting BCAA intake along with optimal nutritional supply to keep the plasma branched-chain metabolites continually close to normal range, protecting the brain from functional disturbances and structural damage (Strauss et al 2010). Abnormalities of immune function and cytokine levels are implicated in the pathophysiology of MSUD. The abnormalities of immune function that have been suggested to occur in MSUD are based on both direct and indirect evidence. For example, De Simone et al (2013) showed that H-BCAA microglial cells exhibit a peculiar phenotype characterized by a partial skewing toward the anti-inflammatory state, with enhanced IL-10 expression and phagocytic activity and increased free radical generation and decreased neuroprotective function. Consistent with this result, we have recently verified that chronic administration of H-BCAA decreases pro-inflammatory cytokine levels in the brains of rats; however, acute administration of H-BCAA increases IL-1β, IL-6, and TNF-α levels and decreases IL-10 levels in the brains of infant rats but not in those of young rats (Rosa et al 2016).
To extend this investigation and better understand the involvement of inflammation in the pathophysiology of MSUD, in the present study, we measured inflammatory markers in plasma from treated patients with the classic form of MSUD. We also investigated whether alterations of those parameters were correlated with plasma leucine, isoleucine, and valine concentrations. We demonstrated that patients with MSUD in treatment with restricted protein diets have elevated levels of pro-inflammatory cytokines [IFN-γ, TNF-α, IL-1β, and IL-6] and cell adhesion molecules [sICAM-1 and sVCAM-1] compared to controls. Our study corroborates previous work published by Mescka et al (2015a, b), which demonstrated that patients with MSUD in treatment with restricted protein diets and without L-carnitine supplementation have increased levels of pro-inflammatory cytokines IL-1β, IL-6, and IFN-ɣ in comparison to controls, and L-carnitine supplementation improved cellular defense against inflammation and oxidative stress. Moreover, we observed that patients with MSUD showed increased ratios of IFN-γ/IL-4, IL-2/IL-4, and IFN-γ/IL-10 in comparison to controls, suggesting a bias toward a Th1 profile in MSUD. When activated, Th cells can differentiate into Th1, Th2, and Th17 cells. These have different immune responses via different patterns of cytokine production; Th1 cells act mainly in macrophage and T cytotoxic cell activation, Th2 cells contribute to B lymphocyte activation, while Th17 cells are responsible for tissue inflammation induction and protection against extracellular pathogens (Striz et al 2014). The bias toward a Th1 profile seen in MSUD reinforces the hypothesis that MSUD is associated with immunological imbalance or dysfunction. The combinations of IFN-γ with TNF-α or IL-1 can strikingly upregulate the expression of two Ig family adhesion molecules: ICAM-1 and VCAM-1. To the best of our knowledge, this is the first report to describe elevated levels of cell adhesion molecules [sICAM-1 and sVCAM-1] in patients with MSUD compared to controls. Thus, we could consider that altered levels of sICAM-1 and sVCAM-1 could be a result of a chronic low-grade inflammation mediated by immune markers that were previously reported to promote Th1 dominance and influence cellular adhesion in MSUD.
Cell adhesion molecules such as sICAM-1 and sVCAM-1 increase T cell infiltration and have been shown to be involved in the development of demyelinating diseases (Zameer and Hoffman 2003; Deem and Cook-Mills 2004; Bullard et al 2007). Ivkovic et al (2017) showed an involvement of sICAM-1 in state and sVCAM-1 in trait vulnerability to mood symptoms. Similarly, another study reported a vulnerability of sICAM-1 to state symptoms in patients with schizophrenia (Schwarz et al 2000). The association of low-grade inflammation and endothelial dysfunction with depression was also demonstrated in a study published by van Dooren et al (2016). In this study, they showed that the levels of TNF-α, CRP, sICAM-1, and low-grade inflammation were associated with depressive disorder. Moreover, studies have shown that the plasma concentrations of sVCAM-1 and sICAM-1 are increased in AD (Rentzos et al 2005; Zuliani et al 2008; Popp et al 2017), suggesting that cell adhesion molecules play an essential role in Ab aggregate-stimulated endothelial-monocyte adhesion (Gonzalez-Velasquez et al 2010). Notably, generalized oedema in the CNS, atrophy of the cerebral hemispheres, spongy degeneration of white matter, and delayed myelinisation, followed by neurocognitive deficits, have been observed in patients with MSUD (Treacy et al 1992; Jan et al 2003; Strauss et al 2010; Shellmer et al 2011; Klee et al 2013; Muelly et al 2013). Taking these findings and the present results together, we suggest that the generalized spongy degeneration of white matter, delayed myelinisation, cognitive impairment, and neuropsychiatric illness observed in patients with MSUD may be mediated by inflammatory responses and endothelial dysfunction.
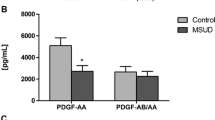
In an attempt to explore the potential clinical correlates of our findings we tested possible correlations between clinical characteristics and inflammatory markers. We found a positive correlation between IL-1β and sICAM-1 levels and metabolic crisis, suggesting that a higher number of metabolic crises may be involved in the immunological imbalance by Th1 profile. Corroborating these results, a study with 35 patients with neonatal-onset MSUD showed that the need for psychological follow-up was significantly associated with the number of lifetime metabolic decompensations (Abi-Warde et al 2017). Although correlation does not necessarily imply causation since we have confounding factors such as recurrent infections, correlation could be evidence for causation, and more work will be needed to explore these possibilities. On the other hand, we observed no correlations between blood amino acid levels and interleukin levels or cell adhesion molecules in this study. Taken together, it may be presumed that leucine and the other BCAAs are not directly associated with pro-inflammatory cytokine production in MSUD. In this line, Mescka et al (2015a, b) suggested that oxidative stress observed in patients with MSUD may be related, at least in part, to the increase in pro-inflammatory cytokine plasma levels, since a positive correlation was found between pro-inflammatory cytokines and MDA values, the main marker of lipid peroxidation, in the plasma of patients with MSUD. Moreover, studies have shown evidence of long-term deficits in neurocognition despite acceptable metabolic control (McLaughlin et al 2013; Vogel et al 2014), suggesting that early lesions to the brain may have long-term consequences (Muelly et al 2013). Additionally, a recent study showed that lower levels of BDNF and PDGF-AA, and higher levels of NCAM and cathepsin D, are not correlated with leucine levels in patients with MSUD under treatment (Scaini et al 2016).
Our results should be interpreted considering their limitations. The small number of patients and healthy controls may be a factor preventing us from finding significant differences in the biomarkers evaluated. Thus, a validation study comprising a larger sample size is needed to confirm the current findings. Moreover, we did not evaluate the impact of inflammatory markers and their relation with cognitive impairment and neuropsychiatric illness in patients with MSUD. Therefore, future longitudinal studies involving inflammatory markers levels related to the Th1 profile may help clarify the role of inflammation in cognitive impairment and psychiatric symptoms among individuals with MSUD.
In conclusion, our study corroborates and expands evidence from previous studies by showing significant changes in inflammatory markers in patients with MSUD. Our data together with previous studies corroborate the hypothesis that immunological mechanisms are involved in the pathogenesis and psychopathology of MSUD, but this process is not directly associated with the BCAA blood levels. A better knowledge of these events may contribute to a more comprehensive understanding of the biological processes in this disease and provide new targets for future treatment strategies to improve the quality of life of MSUD patients.
References
Abi-Warde MT, Roda C, Arnoux JB et al (2017) Long-term metabolic follow-up and clinical outcome of 35 patients with maple syrup urine disease. J Inherit Metab Dis 40:783–792
Amaral AU, Leipnitz G, Fernandes CG, Seminotti B, Schuck PF, Wajner M (2010) Alpha-ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res 1324:75–84
Amor S, Puentes F, Baker D, van der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology 129:154–169
Amor S, Peferoen LA, Vogel DY et al (2014) Inflammation in neurodegenerative diseases--an update. Immunology 142:151–166
Araujo P, Wassermann GF, Tallini K et al (2001) Reduction of large neutral amino acid levels in plasma and brain of hyperleucinemic rats. Neurochem Int 38:529–537
Barschak AG, Sitta A, Deon M et al (2008) Oxidative stress in plasma from maple syrup urine disease patients during treatment. Metab Brain Dis 23:71–80
Barschak AG, Sitta A, Deon M et al (2009) Amino acids levels and lipid peroxidation in maple syrup urine disease patients. Clin Biochem 42:462–466
Bridi R, Araldi J, Sgarbi MB et al (2003) Induction of oxidative stress in rat brain by the metabolites accumulating in maple syrup urine disease. Int J Dev Neurosci Off J Int Soc Dev Neurosci 21:327–332
Bridi R, Braun CA, Zorzi GK et al (2005) Alpha-keto acids accumulating in maple syrup urine disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab Brain Dis 20:155–167
Bullard DC, Hu X, Schoeb TR, Collins RG, Beaudet AL, Barnum SR (2007) Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J Immunol 178:851–857
Chuang DT, Shih VE (2001) Maple syrup urine disease (branched-chain ketoaciduria). In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 1971–2005
Crome L, Dutton G, Ross CF (1961) Maple syrup urine disease. J Pathol Bacteriol 81:379–384
Cunningham C, Hennessy E (2015) Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther 7:33
Dancis J, Hutzler J, Cox RP (1977) Maple syrup urine disease: branched-chain keto acid decarboxylation in fibroblasts as measured with amino acids and keto acids. Am J Hum Genet 29:272–279
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
De Simone R, Vissicchio F, Mingarelli C et al (2013) Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim Biophys Acta 1832:650–659
Deem TL, Cook-Mills JM (2004) Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood 104:2385–2393
Deon M, Sitta A, Faverzani JL et al (2015) Urinary biomarkers of oxidative stress and plasmatic inflammatory profile in phenylketonuric treated patients. Int J Dev Neurosci Off J Int Soc Dev Neurosci 47:259–265
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010) Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68:748–754
Fakhoury M (2015) Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis 15:63–69
Fingerhut R (2009) Recall rate and positive predictive value of MSUD screening is not influenced by hydroxyproline. Eur J Pediatr 168:599–604
Flaschker N, Feyen O, Fend S, Simon E, Schadewaldt P, Wendel U (2007) Description of the mutations in 15 subjects with variant forms of maple syrup urine disease. J Inherit Metab Dis 30:903–909
Funchal C, Latini A, Jacques-Silva MC et al (2006a) Morphological alterations and induction of oxidative stress in glial cells caused by the branched-chain alpha-keto acids accumulating in maple syrup urine disease. Neurochem Int 49:640–650
Funchal C, Schuck PF, Santos AQ et al (2006b) Creatine and antioxidant treatment prevent the inhibition of creatine kinase activity and the morphological alterations of C6 glioma cells induced by the branched-chain alpha-keto acids accumulating in maple syrup urine disease. Cell Mol Neurobiol 26:67–79
Gonzalez-Velasquez FJ, Reed JW, Fuseler JW et al (2010) Soluble amyloid-β protein aggregates induce nuclear factor-κB mediated upregulation of adhesion molecule expression to stimulate brain endothelium for monocyte adhesion. J Adhes Sci Technol 24:2105–2126
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009) Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66:407–414
Henneke M, Flaschker N, Helbling C et al (2003) Identification of twelve novel mutations in patients with classic and variant forms of maple syrup urine disease. Hum Mutat 22:417
Imtiaz F, Al-Mostafa A, Allam R, Ramzan K, Al-Tassan N, Tahir AI, Al-Numair NS, Al-Hamed MH, Al-Hassnan Z, Al-Owain M, Al-Zaidan H, Al-Amoudi M, Qari A, Balobaid A, Al-Sayed M (2017) Twenty novel mutations in BCKDHA, BCKDHB and DBT genes in a cohort of 52 Saudi Arabian patients with maple syrup urine disease. Mol Genet Metab Rep 11:17-23
Ivkovic M, Pantovic-Stefanovic M, Petronijevic N et al (2017) Predictive value of sICAM-1 and sVCAM-1 as biomarkers of affective temperaments in healthy young adults. J Affect Disord 207:47–52
Jan W, Zimmerman RA, Wang ZJ, Berry GT, Kaplan PB, Kaye EM (2003) MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology 45:393–399
Joseph MH, Marsden CA (1986) Amino acids and small peptides) In: Lim CF (ed) HPLC of small peptides. IRL Press, Oxford, pp 13–27
Jouvet P, Rustin P, Felderhoff U et al (1998) Maple syrup urine disease metabolites induce apoptosis in neural cells without cytochrome c release or changes in mitochondrial membrane potential. Biochem Soc Trans 26:S341
Jouvet P, Kozma M, Mehmet H (2000a) Primary human fibroblasts from a maple syrup urine disease patient undergo apoptosis following exposure to physiological concentrations of branched chain amino acids. Ann N Y Acad Sci 926:116–121
Jouvet P, Rustin P, Taylor DL et al (2000b) Branched chain amino acids induce apoptosis in neural cells without mitochondrial membrane depolarization or cytochrome c release: implications for neurological impairment associated with maple syrup urine disease. Mol Biol Cell 11:1919–1932
Klee D, Thimm E, Wittsack HJ et al (2013) Structural white matter changes in adolescents and young adults with maple syrup urine disease. J Inherit Metab Dis 36:945–953
Laske C, Zank M, Klein R et al (2008) Autoantibody reactivity in serum of patients with major depression, schizophrenia and healthy controls. Psychiatry Res 158:83–86
Mackenzie DY, Woolf LI (1959) Maple syrup urine disease; an inborn error of the metabolism of valine, leucine, and isoleucine associated with gross mental deficiency. Br Med J 1:90–91
McLaughlin PM, Hinshaw J, Stringer AY (2013) Maple syrup urine disease (MSUD): a case with long-term follow-up after liver transplantation. Clin Neuropsychol 27:1199–1217
Menkes JH (1959) Maple syrup disease; isolation and identification of organic acids in the urine. Pediatrics 23:348–353
Mescka CP, Wayhs CA, Vanzin CS et al (2013) Protein and lipid damage in maple syrup urine disease patients: l-carnitine effect. Int J Dev Neurosci Off J Int Soc Dev Neurosci 31:21–24
Mescka CP, Guerreiro G, Donida B et al (2015a) Investigation of inflammatory profile in MSUD patients: benefit of L-carnitine supplementation. Metab Brain Dis 30:1167–1174
Mescka CP, Guerreiro G, Hammerschmidt T et al (2015b) L-carnitine supplementation decreases DNA damage in treated MSUD patients. Mutat Res 775:43–47
Muelly ER, Moore GJ, Bunce SC et al (2013) Biochemical correlates of neuropsychiatric illness in maple syrup urine disease. J Clin Invest 123:1809–1820
Palta P, Xue QL, Deal JA, Fried LP, Walston JD, Carlson MC (2015) Interleukin-6 and C-reactive protein levels and 9-year cognitive decline in community-dwelling older women: the women’s health and aging study II. J Gerontol A Biol Sci Med Sci 70:873–878
Popp J, Oikonomidi A, Tautvydaite D et al (2017) Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav Immun 62:203–211
Quental S, Vilarinho L, Martins E et al (2010) Incidence of maple syrup urine disease in Portugal. Mol Genet Metab 100:385–387
Raison CL, Miller AH (2011) Is depression an inflammatory disorder? Curr Psychiatry Rep 13:467–475
Rentzos M, Michalopoulou M, Nikolaou C et al (2005) The role of soluble intercellular adhesion molecules in neurodegenerative disorders. J Neurol Sci 228:129–135
Ribeiro CA, Sgaravatti AM, Rosa RB et al (2008) Inhibition of brain energy metabolism by the branched-chain amino acids accumulating in maple syrup urine disease. Neurochem Res 33:114–124
Ribeiro LR, Della-Pace ID, de Oliveira Ferreira AP et al (2013) Chronic administration of methylmalonate on young rats alters neuroinflammatory markers and spatial memory. Immunobiology 218:1175–1183
Rosa L, Scaini G, Furlanetto CB et al (2016) Administration of branched-chain amino acids alters the balance between pro-inflammatory and anti-inflammatory cytokines. Int J Dev Neurosci Off J Int Soc Dev Neurosci 48:24–30
Scaini G, de Rochi N, Jeremias IC et al (2012) Evaluation of acetylcholinesterase in an animal model of maple syrup urine disease. Mol Neurobiol 45:279–286
Scaini G, Comim CM, Oliveira GM et al (2013a) Chronic administration of branched-chain amino acids impairs spatial memory and increases brain-derived neurotrophic factor in a rat model. J Inherit Metab Dis 36:721–730
Scaini G, Mello-Santos LM, Furlanetto CB et al (2013b) Acute and chronic administration of the branched-chain amino acids decreases nerve growth factor in rat hippocampus. Mol Neurobiol 48:581–589
Scaini G, Morais MO, Furlanetto CB et al (2015) Acute Administration of Branched-Chain Amino Acids Increases the pro-BDNF/Total-BDNF ratio in the rat brain. Neurochem Res 40:885–893
Scaini G, Tonon T, de Souza CF et al (2016) Serum markers of neurodegeneration in maple syrup urine disease. Mol Neurobiol 54:5709–5719
Schmidt R, Schmidt H, Curb JD, Masaki K, White LR, Launer LJ (2002) Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia aging study. Ann Neurol 52:168–174
Schwarz MJ, Riedel M, Ackenheil M, Muller N (2000) Decreased levels of soluble intercellular adhesion molecule-1 (sICAM-1) in unmedicated and medicated schizophrenic patients. Biol Psychiatry 47:29–33
Seminotti B, Amaral AU, Ribeiro RT et al (2016) Oxidative stress, disrupted energy metabolism, and altered signaling pathways in Glutaryl-CoA dehydrogenase knockout mice: potential implications of quinolinic acid toxicity in the neuropathology of glutaric acidemia type I. Mol Neurobiol 53:6459–6475
Sgaravatti AM, Rosa RB, Schuck PF et al (2003) Inhibition of brain energy metabolism by the alpha-keto acids accumulating in maple syrup urine disease. Biochim Biophys Acta 1639:232–238
Shellmer DA, DeVito DA, Dew MA et al (2011) Cognitive and adaptive functioning after liver transplantation for maple syrup urine disease: a case series. Pediatr Transplant 15:58–64
Silberman J, Dancis J, Feigin I (1961) Neuropathological observations in maple syrup urine disease: branched-chain ketoaciduria. Arch Neurol 5:351–363
Simon E, Fingerhut R, Baumkotter J, Konstantopoulou V, Ratschmann R, Wendel U (2006) Maple syrup urine disease: favourable effect of early diagnosis by newborn screening on the neonatal course of the disease. J Inherit Metab Dis 29:532–537
Singh-Manoux A, Dugravot A, Brunner E et al (2014) Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology 83:486–493
Sitta A, Ribas GS, Mescka CP, Barschak AG, Wajner M, Vargas CR (2014) Neurological damage in MSUD: the role of oxidative stress. Cell Mol Neurobiol 34:157–165
Strauss KA, Wardley B, Robinson D et al (2010) Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab 99:333–345
Striz I, Brabcova E, Kolesar L, Sekerkova A (2014) Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond) 126(9):593–612
Tavares RG, Santos CE, Tasca CI, Wajner M, Souza DO, Dutra-Filho CS (2000) Inhibition of glutamate uptake into synaptic vesicles of rat brain by the metabolites accumulating in maple syrup urine disease. J Neurol Sci 181:44–49
Treacy E, Clow CL, Reade TR, Chitayat D, Mamer OA, Scriver CR (1992) Maple syrup urine disease: interrelations between branched-chain amino-, oxo- and hydroxyacids; implications for treatment; associations with CNS dysmyelination. J Inherit Metab Dis 15:121–135
van Dooren FE, Schram MT, Schalkwijk CG et al (2016) Associations of low grade inflammation and endothelial dysfunction with depression - the Maastricht study. Brain Behav Immun 56:390–396
Victora CG, Huttly SR, Fuchs SC, Olinto MT (1997) The role of conceptual frameworks in epidemiological analysis: a hierarchical approach. Int J Epidemiol 26:224–227
Vogel KR, Arning E, Wasek BL, McPherson S, Bottiglieri T, Gibson KM (2014) Brain-blood amino acid correlates following protein restriction in murine maple syrup urine disease. Orphanet J Rare Dis 9:73
Wajner M, Vargas CR (1999) Reduction of plasma concentrations of large neutral amino acids in patients with maple syrup urine disease during crises. Arch Dis Child 80:579
Wajner M, Coelho DM, Barschak AG et al (2000) Reduction of large neutral amino acid concentrations in plasma and CSF of patients with maple syrup urine disease during crises. J Inherit Metab Dis 23:505–512
Wajner A, Burger C, Dutra-Filho CS, Wajner M, de Souza Wyse AT, Wannmacher CM (2007) Synaptic plasma membrane Na(+), K (+)-ATPase activity is significantly reduced by the alpha-keto acids accumulating in maple syrup urine disease in rat cerebral cortex. Metab Brain Dis 22:77–88
Yaffe K, Lindquist K, Penninx BW et al (2003) Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology 61:76–80
Zameer A, Hoffman SA (2003) Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol 142:67–74
Zielke HR, Zielke CL, Baab PJ, Collins RM (2002) Large neutral amino acids auto exchange when infused by microdialysis into the rat brain: implication for maple syrup urine disease and phenylketonuria. Neurochem Int 40:347–354
Zinnanti WJ, Lazovic J, Griffin K et al (2009) Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain J Neurol 132:903–918
Zuliani G, Cavalieri M, Galvani M et al (2008) Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J Neurol Sci 272:164–170
Acknowledgements
Laboratory of Bioenergetics (Brazil) is one of the centres of the National Institute for Molecular Medicine (INCT-MM) and one of the members of the Center of Excellence in Applied Neurosciences of Santa Catarina (NENASC). This research was supported by grants from CNPq (402047/2010-9), FAPESC, and UNESC. We are also grateful to INAGEMP (Brazilian National Institute of Population Medical Genetics) for the support provided through grants FAPERGS #17/2551.0000521.0, CNPq #465549/2014-4 and CAPES #88887.136366/2017-00. The authors acknowledge all the members of the Brazilian MSUD Network, especially the LAM- SGM/HCPA group for helping us with the quantitative amino acid analysis as well as Dr. Kevin Strauss and Dr. Erik Puffenberger (Clinical for Special Children, Pennsylvania-USA) for helping us with the alloisoleucine measurement. The authors declare that they have not had any financial, personal or other relationships that have influenced the work.
Funding
This research was supported by grants from CNPq (402047/2010-9), FAPESC, and UNESC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
G. Scaini, T. Tonon, C. F. Moura de Souza, P. F. Schuck, G. C. Ferreira, J. Quevedo, J. S. Neto, T. Amorim, J. S. Camelo Jr, A. V. B. Margutti, R. Tresbach, F. Sperb-Ludwig, R. Boy, P. F. V. de Medeiros, I. V. D. Schwartz, E. L. Streck declare that they have no conflict of interest.
Additional information
Communicated by: Eva Morava
Electronic supplementary material
ESM 1
(DOCX 21 kb)
Rights and permissions
About this article

Cite this article
Scaini, G., Tonon, T., Moura de Souza, C.F. et al. Evaluation of plasma biomarkers of inflammation in patients with maple syrup urine disease. J Inherit Metab Dis 41, 631–640 (2018). https://doi.org/10.1007/s10545-018-0188-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-018-0188-x