
Abstract
Objectives
To develop an efficient gene-targeting platform in an excellent itaconic acid producing strain Aspergillus terreus CICC40205.
Results
The frequency of homologous recombination was improved by deleting the ku80 gene. A nutritional transformation system based on the bidirectionally selectable marker, pyrG An , was established in the ku80-/pyrG-double mutant which is convenient for following marker rescue. The modified Cre/loxP recombination system was applied for the excision of the pyrG An marker by directly introducing Cre recombinase into the protoplasts.
Conclusions
This gene-targeting system is an efficient platform for sequential and multiple genetic modifications in A. terreus and is conducive to study biosynthesis mechanisms and to improve the production ability of itaconic acid and other products.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aspergillus terreus is a ubiquitously distributed saprobiotic mold fungus but can cause life-threatening invasive aspergillosis in immunocompromised patients (Slesiona et al. 2012). Importantly, A. terreus is a significant species with biotechnological and medical values and has been commercially developed as an excellent producer for lovastatin (e.g. Merck’s Mevacor) (Barrios-Gonzalez and Miranda 2010) and itaconic acid (Klement and Buchs 2013). Lovastatin is an efficient cholesterol-lowering agent, with worldwide sales topping $10 billion annually (Barrios-Gonzalez and Miranda 2010). Itaconic acid is widely used as a monomer or co-monomer in manufacturing plastics, resins etc. (Klement and Buchs 2013).
With the rapid increase of the fungal omics data, genetic and metabolic engineering approaches have become powerful for functional characterization of the genes and strain improvement. Gene targeting is one of the most important methods for genetic engineering. However, as with other Aspergillus spp, two limiting factors hamper the studies towards genetic engineering of A. terreus: (1) low frequency of homologous recombination (HR); (2) limited suitable resistance markers. Considering that a large number of genes will be functionally analyzed and manipulated, it is important to develop an efficient platform facilitating this process.
In filamentous fungi, integration of a DNA fragment into the genome is mainly mediated by non-homologous end joining (NHEJ) pathways, resulting in low frequency in gene-targeting. Hence, a multitude of candidate transformants have usually to be analyzed in order to identify the correct one, which makes fungal transformation laborious, tedious and time-consuming. The expression cassette of target gene is randomly integrated into the genome which is not ideal for the comparison of phenotypes between different mutants (Huang et al. 2014a). However, exogenous NDA can be mainly integrated into the genome through HR in the NHEJ-pathway defective mutant (Ishibashi et al. 2006). The frequency of gene-targeting can be significantly improved by deleting the genes ku70, ku80 or lig4, the key components of the NHEJ-pathway (Ishibashi et al. 2006; Mizutani et al. 2008; Honda et al. 2011). The NHEJ-pathway defective mutants have been used as the parental strains to study the biosynthesis of secondary metabolites in A. terreus (Gressler et al. 2011; Guo et al. 2013).
Selectable marker genes are also essential for genetic transformation. The modification of numerous genes by individual gene targeting with different selectable markers, however, is not appropriate for A. terreus being grown commercially because of the high costs involved. Moreover, mutants with resistance markers when used on an industrial scale may also raise public concerns on biosafety. Therefore, recyclable marker modules that allow repetitive rounds of transformations by marker rescue are essential for sequential genetic modifications and environmental security. Site-specific recombination is an important molecular tool for functional genetic studies and has been applied for marker rescue in various microorganisms, including Aspergillus sp. (Forment et al. 2006; Florea et al. 2009; Kopke et al. 2010). In the traditional strategy, the recombinase expression cassette should be introduced into the cells and induced (Florea et al. 2009; Kopke et al. 2010). Mizutani et al. (2012) investigated a simple method to use modified Cre/loxP recombination system in Aspergillus oryzae by directly introducing Cre recombinase into protoplast.
Although there have been some examples of improving the itaconic acid accumulation through genetic engineering (Lin et al. 2004; Tevz et al. 2010), just overexpressing the related genes individually in an industrially-used A. terreus strain could not improving the itaconic acid productivity significantly (Huang et al. 2014b). So the genetic engineering of multiple genes with multiple modification approaches would be expected. Here, with a view to studying the regulation mechanisms of itaconic acid biosynthesis and improve the ability of itaconic acid production, we developed an efficient marker-free gene-targeting system with high HR frequency and friendly marker rescue in an excellent itaconic acid production strain A. terreus CICC40205. The system should be also valuable to explore and engineer other A. terreus strains producing other products.
Materials and methods
Materials
Cre recombinase (M0298L) was obtained from NEB (USA). Lysing enzymes (1412) and cellulose (C1184) were from Sigma-Aldrich (USA), and the snailase was from Sangon (Shanghai, China). The DIG High Prime DNA Labeling and Detection Starter Kit I was purchased from Roche.
Strains, medium, and cultivation conditions
A. terreus strains used in this project are listed in Table 1. A. terreus CICC 40205 was obtained from China Centre of Industrial Culture Collection. The spores were cultured on AMMB agar [Aspergillus minimal medium (AMM, http://www.fgsc.net/methods/anidmed.html) with 5 g wheat bran l−1] at 32 °C for 7 days. Further cultivation was carried out in 500 ml non-baffled shake-flasks containing 55 ml itaconic acid production medium (IPM) on a rotary shaker at 200 rpm and 37 °C (Huang et al. 2014c).
Construction of the ku80 deletion mutant At-∆ku80
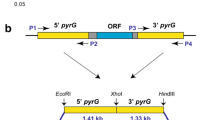
The primers used in this study are listed in Supplementary Table 1. The ku80 (ATEG_06919) was deleted using the split-marker approach. The flanking regions of ku80 with length of about 1.5 kb were amplified from the genome of A. terreus CICC40205 using primer pairs Uku80-F/Uku80-R and Dku80-F/Dku80-R respectively, and then fused with selectable marker ptrA by fusion PCR. Two hybrid split-marker constructs, ku80-A containing the 5′-flank of ku80 and 3/4 of the ptrA from the 5′-end, and ku80-B containing the 3′-flank of ku80 and 1/2 of the ptrA from the 3′-end, were amplified from the fusion PCR products using primer pairs Cku80-F/ptrA-744R and ptrA-177F/Cku80-R respectively (Fig. 1a).

Schematic model of the construction of mutants At-∆ku80 and At-∆pyrG. a The deletion of ku80 gene using split-marker approach. b The deletion of pyrG gene in At-∆ku80 strain
2 μg ku80-A and 1.5 μg ku80-B constructs were transformed together into A. terreus CICC40205 using the protoplast-PEG method as described (Blumhoff et al. 2013). The transformants were screened on AMMS (AMM with 1.2 M sorbitol) agar plates supplemented with 0.1 mg pyrithiamine (PT) l−1, and purified by single spore isolation. The genotypes of ∆ku80::ptrA putative mutants were examined by genomic PCR using primer pairs ptrA-F/ptrA-R, Uku80-F/ptrA-R, ptrA-F/Dku80-R, and Sku80-F/Sku80-R. The genomic DNA of the candidate mutants were digested with BamHI respectively, and used for Southern blot analysis. The probe ptrA-S used for hybridization was obtained by PCR with primer ptrA-F171/ptrA–R744.
Construction of the uracil auxotrophic mutant At-∆pyrG
The upstream and downstream sequences of pyrG (ATEG_09675) were amplified using primer pairs UpyrG-F/UpyrG-R and DpyrG-F/DpyrG-R respectively, and joined together using fusion PCR. The gene-targeting construct was amplified using primers CpyrG-F/CpyrG-R and transformed into At-∆ku80 mutant (Fig. 1b). The transformants were selected on AMMS plates with 10 mM uracil and 0.5 g 5-fluoroorotic acid (5-FOA) l−1, and purified by single spore isolation. The pyrG-deficient candidates were incubated on the AMM plate without uracil to test the uracil dependence. The genotypes were confirmed by genomic PCR using primer pairs UpyrG-F/D-pyrG-R and pyrG-F/pyrG-R respectively.
Complementation of At-∆pyrG mutant with pyrG An of Aspergillus niger
The DNA fragment comprising loxP-pyrG An -loxP, PalcA-sgfp expression cassette, and the flank sequences of ku80 was amplified from plasmid pXH106 (Supplementary Fig. 1), and transferred into the protoplasts of At-∆pyrG mutant. The transformants were selected on AMMS plates and purified by single spore isolation. The genotypes were verified by PCR analysis using primer pairs pyrGAn-F/TtrpC-R, Uku80-F/pyrGAn-R, pyrGAn-F/Dku80-R, ptrA-F/ptrA-R respectively.
Direct introduction of Cre recombinase into At-loxP-pyrGAn cells for maker rescue
Cre recombinase was introduced into the At-loxP-pyrGAn cells as described previously with some modifications (Mizutani et al. 2012). The protoplasts of At-loxP-pyrGAn were prepared and adjusted to 5 × 108 cells ml−1 in STC buffer. 100 μl protoplast solution was mixed well with 15 μl PSTC buffer, 10 μl Cre recombinase, and 4 μg plasmid pXH-pyrGAn (Supplementary Fig. 1). The mixture was incubated on ice for 30 min and 1 ml PSTC was added. The transformants were selected on AMMS plates with 10 mM uracil and 0.5 g 5-FOA l−1, and purified by subculturing for times. The uracil dependence of the transformants were confirmed by incubating on the AMM plate. The genotypes were confirmed by PCR using primer pair Uku80-F/TtrpC-R, and the PCR product were sequenced using primer s-ku80-F.
Characterization of the mutants
To measure the HR frequency in the ku80 deletion mutant, the ATEG_04633 gene was deleted by gene-targeting in mutant strain At-∆ku80. The gene-targeting element of ATEG_04633 was constructed as follows: The selectable marker hph was amplified from pG3H (Huang et al. 2014c) using primer hph-F/hph-R. The flanking regions of ATEG_04633 were amplified from the genome of A. terreus CICC40205 using primer pairs U4633-F/U4633-R and D4633-F/D4633-R respectively, and then fused with hph by fusion PCR. The gene-targeting element was amplified by primers C4633-F/C4633-R and transformed into At-∆ku80 mutant. The candidates were selected on potato/dextrose/agar plates with 1.2 M sorbitol and 100 mg hygromycin B l−1, and analyzed by PCR. To examine the uracil auxotrophy, 5-FOA and PT resistant of mutants, the WT, At-∆ku80, At-∆pyrG, At-loxP-pyrGAn, At-loxP-∆pyrGAn strains were incubated on AMM plates for 4 days, AMM plates supplemented 0.1 mg PT l−1 and 10 mM uracil for 7 days, and AMM plates supplemented 10 mM 5-FOA and 10 mM uracil for 7 days. The itaconic acid productivities and growth rates of the WT, At-∆ku80, At-loxP-pyrGAn mutants were investigated by cultivation in shake flasks as described previously (Huang et al. 2014c).
Results and discussion
Deletion of ku80 by the split-marker approach
To avoid the ectopic integrations of the DNA fragment containing the intact ptrA marker, the split-marker approach was adopted to disrupt the ku80 gene (ATEG_06919) of A. terreus CICC40205 as illustrated in Fig. 1a (Fu et al. 2006; Aragona and Valente 2015). The split-marker constructs ku80-A and ku80-B which contain a 597-bp length overlap region located in the coding sequence of the ptrA marker were transformed into A. terreus CICC40205. Three transformants with PT resistance were confirmed by genomic PCR, and showing that the ku80 were deleted completely in transformants #1 and #2 (Supplementary Fig. 2a).
The DNA fragments can yield unexpected ectopic integrations because of the domination of NHEJ, even in the transformants with deletion of ku80. These unexpected integrations may cause unpredictable influence on the strain. To identify the completely correct deletion of ∆ku80 mutant, the probe ptrA-S was designed within the overlap region of two split-marker constructs and applied in the southern blot analysis. The result showed that only the band of correct integration (4.4 kb) appeared in the lane of transformant 2# (Supplementary Fig. 2b), which demonstrated that the transformant 2# with ku80 deletion is the anticipated and exact At-∆ku80 mutant without any ectopic integration.
Characterization of the ku80 deletion strain At-∆ku80
To evaluate the influences of ku80 deletion, the phenotypic characteristics of At-∆ku80 were assessed. In comparison to the wild-type, At-∆ku80 mutant did not show any significantly difference in the growth rates, conidiation or pigmentation on AMMB agar plates (Fig. 2a). These results were similar to those of A. fumigatus and A. niger (Krappmann et al. 2006; Honda et al. 2011). Furthermore, no deviations in itaconic acid productivity and vegetative growth were observed in the itaconic acid fermentation, indicating that the deletion of the ku80 gene in A. terreus CICC40205 did not have adverse effect on itaconate production (Fig. 2b). Similar results were also observed in the citric acid producing A. niger strain (Honda et al. 2011).

Comparison of the mutants used in this study. a Comparison of cell growth on AMMB plates. WT, AT-∆ku80 and At-loxP-pyrGAn strains were compared in the growth rate and spore morphology. b Comparison of itaconic acid production of the WT, AT-∆ku80 and At-loxP-pyrGAn strains. The conidia were inoculated in IPM medium and cultivated on a rotary shaker at 37 °C for 4 days. Three independent experiments were performed for each sample. The dry weight of biomass and itaconic acid titers were measured
To test the HR frequency of the mutant At-∆ku80, the pdc gene (ATEG_04633) encoding pyruvate decarboxylase was targeted for the deletion in ku80-deficient mutant, At-∆ku80. Pyruvate decarboxylase is proposed as the mainly competitive enzyme for the metabolism of pyruvate, the precursor of itaconic acid biosynthesis. Although the fermentation result showed that the deletion of the pdc gene did not benefit the production of itaconate (data not shown), the HR frequency of the mutant At-∆ku80 during the experiment of pdc deletion was significantly improved. In 19 out of 20 transformants, the gene targeting constructs were correctly integrated at the pdc loci through HR (Fig. 3). In wild type the HR frequency is very low, about 10 % (date not shown). These results clearly showed that the frequency of homologous integration had been improved to 95 % in the ku80-deficient mutant, and that we generated an efficient recipient for gene-targeting.

Deletion of ATEG_04633 gene in A. terreus At-∆ku80 strain. a Strategy of deleting ATEG_04633 gene in At-∆ku80 strain. The genotypes of 20 transformants were verified by genomic PCR using primer pairs hph-F/hph-R (b), U4633-F/hph-R (c), hph-F/D4633-R (d), and 4633-F/4633-R (e) respectively. Lane M 1 kb DNA marker; lane c, the negative control, At-∆ku80 strain; 1–20, the transformants
Construction of a bidirectional selection system based on pyrG
The bidirectional selection system based on nutritional marker pyrG was successfully applied in several kinds of filamentous fungi (Mattern et al. 1987; Kanamasa et al. 2003; Wang et al. 2010; Guo et al. 2013). In this study, the uracil auxotrophy strain At-∆pyrG was generated by deleting the whole pyrG gene in mutant At-∆ku80 (Fig. 1b). Two candidates which could not grow on the AMM plate without uracil were characterized by genomic PCR, which confirmed that the pyrG were deleted by HR in both two candidates (Supplementary Fig. 3). Besides, we also found that the mutant At-∆pyrG grew slowly than At-∆ku80 on the AMM plate containing PT and uracil, indicating that the deletion of pyrG gene had adverse effect on the growth of A. terreus even on the medium containing uracil (Fig. 4d).

Strategy for the transformation and marker rescue based on pyrG An marker. a Complementation of At-∆pyrG strain with pyrG An . The uracil auxotroph At-∆pyrG strain was transformed to uracil prototrophy by complementing the loxP-pyrG An -loxP marker at the ku80 loci, and the ptrA marker was replaced. b Schematic representation of pyrG An marker rescue from the At-loxP-pyrGAn strain. The pyrG An marker was rescued using the Cre/loxP system by directly introducing Cre into the protoplast cells. The pyrG An marker located between the two loxP sites was excised and only one loxP site was left. c Genomic PCR analysis of pyrG An marker rescue from the At-loxP-pyrGAn mutant using primers Uku80-F/TtrpC-R. C, At-loxP-pyrGAn strain; 1–8, the At-loxP-∆pyrGAn mutants with pyrG An marker rescue. d The phenotype of the mutants on different agar plates. The WT, At-∆ku80, At-∆pyrG, At-loxP-pyrGAn, At-loxP-∆pyrGAn strains were incubated on AMM plates for 4 days, AMM plates supplemented 0.1 mg l−1 pyrithiamine and 10 mM uracil for 7 days, and AMM plates supplemented 10 mM 5-fluoroorotic acid (5-FOA) and 10 mM uracil for 7 days
To confirm that the At-∆pyrG strain can be used as a recipient in the transformation using the pyrG gene as a selectable marker. The functional pyrG expression cassette of A. niger Co827, pyrG An , was cloned and used for the complementation test of mutant At-∆pyrG (Fig. 4a). The result showed that it transformed At-∆pyrG to uracil prototrophy (Fig. 4d). Importantly, the At-loxP-pyrGAn grew normally as the At-∆ku80 and WT strains on the plates without uracil, and showed similar itaconic acid production level in the fermentation test (Fig. 2). Thus, these results demonstrated that the adverse effects caused by pyrG deficiency could be completely repaired by complementing the pyrG An marker.
In addition the PCR analysis result confirmed that the loxP-pyrG An -loxP selectable marker was integrated at ku80 loci of At-∆pyrG mutant and replaced the ptrA gene through HR in 20 out of 22 transformants (Supplementary Fig. 4). This information further demonstrated that the gene-targeting frequency had been improved from10 to 91 % by deleting the ku80.
Elimination of pyrG An marker by directly introducing Cre recombinase into the cells
The mutant At-loxP-pyrGAn integrating the loxP-pyrG An -loxP cassette was used as the parental strains to test the pyrG An marker elimination. The protoplasts of At-loxP-pyrGAn were incubated with Cre recombinase and pXH-pyrGAn plasmid harboring the functional pyrG of A. niger on ice, and selected on AMMS plates with 10 mM uracil and 0.5 g 5-FOA l−1. More than 100 transformants were obtained, and eight of them were selected randomly and analyzed by genomic PCR with primer pairs as indicated (Fig. 4b). A 3.3-kb length fragment which could be amplified only after excision of pyrG An was identified in all of the eight transformants, indicating the pyrG An gene was rescued successfully in these transformants, designated as At-loxP-∆pyrGAn. While a 4.7-kb length fragment could be obtained when if the pyrG An cassette was still present in the genome, such as the mutant At-loxP-pyrGAn (Fig. 4c). According to the DNA sequencing result of the 3.3 kb fragment, the loxP-pyrG An -loxP marker located between the ku80-UP and PalcA disappeared and only one loxP site was left (Supplementary Fig. 5): that is, two loxP sites were exactly cut and ligated together by the Cre recombinase, and the pyrG An marker was simultaneously cleanly excised. The phenotype test showed that the At-loxP-∆pyrGAn mutant can grow on the AMM plates containing 5-FOA and uracil but not on AMM plates without uracil (Fig. 4d). Therefore the resulting At-loxP-∆pyrGAn mutant strain was transformable using pyrG An marker again. The possible positive occurrence of marker rescue in the transformants was 100 % which was consistent with the results observed in A. oryzae (Mizutani et al. 2012).
In the conventional marker rescue method based on the site-specific recombination, the recombinase was expressed intracellularly by introducing an inducible recombinase expression cassette into the cell. This is a complex and time-consuming process (Krappmann et al. 2005; Forment et al. 2006; Florea et al. 2009). In this modified Cre/loxP system, the Cre recombinase, which is commercially available, was directly introduced into the cells using DNA as a carrier, and the inducible promoter or self-replicating plasmid is no longer needed, and the process of plasmid loss is also eliminated (Mizutani et al. 2012). In this study, a convenient and effective method was introduced into A. terreus. Compared with the dominant marker, the bidirectional selectable marker pyrG An brings great convenience for marker extinction because the marker-free mutants can be screened out efficiently and easily on the AMM plates supplemented with 5-FOA and uracil. To avoid the unexpected ectopic integrations of the carrier DNA, the pXH-pyrGAn plasmid containing the functional pyrG An expression cassette was used as the carrier DNA, because the transformants could not grow healthily on the selection plate AMMS while the pyrG An DNA was integrated into the genome. In addition to the recyclable marker modules based on the site-specific recombination, the marker gene flanked with two direct repeats could also been self-excised by the internal HR of two repeats. The NHEJ-deficient parental strain and bidirectional selectable marker are the critical parts for this method (Maruyama and Kitamoto 2008; Tani et al. 2013).
In conclusion, an efficient gene-targeting platform was developed in an itaconic acid-producing A. terreus strain. This system possesses three prominent features: a chassis cell with high HR frequency, a transformation system based on bidirectionally selectable marker pyrG, and an efficient module for selectable marker rescue by directly introducing Cre recombinase into cells. This gene-targeting system is an efficient platform for multiple genetic modifications in A. terreus, which is significantly conducive to study biosynthesis mechanisms and improve the production ability of itaconic acid, lovastatin, and other high-valued compounds by genetic engineering A. terreus.
References
Aragona M, Valente MT (2015) Genetic transformation of the tomato pathogen Pyrenochaeta lycopersici allowed gene knockout using a split-marker approach. Curr Genet 61:211–220
Barrios-Gonzalez J, Miranda RU (2010) Biotechnological production and applications of statins. Appl Microbiol Biotechnol 85:869–883
Blumhoff M, Steiger MG, Marx H, Mattanovich D, Sauer M (2013) Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl Microbiol Biotechnol 97:259–267
Florea S, Andreeva K, Machado C, Mirabito PM, Schardl CL (2009) Elimination of marker genes from transformed filamentous fungi by unselected transient transfection with a Cre-expressing plasmid. Fungal Genet Biol 46:721–730
Forment JV, Ramon D, MacCabe AP (2006) Consecutive gene deletions in Aspergillus nidulans: application of the Cre/loxP system. Curr Genet 50:217–224
Fu J, Hettler E, Wickes BL (2006) Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genet Biol 43:200–212
Gressler M, Zaehle C, Scherlach K, Hertweck C, Brock M (2011) Multifactorial induction of an orphan PKS-NRPS gene cluster in Aspergillus terreus. Chem Biol 18:198–209
Guo CJ, Knox BP, Sanchez JF, Chiang YM, Bruno KS, Wang CCC (2013) Application of an efficient gene targeting system linking secondary metabolites to their biosynthetic genes in Aspergillus terreus. Org Lett 15:3562–3565
Honda Y, Kobayashi K, Kirimura K (2011) Increases in gene-targeting frequencies due to disruption of kuea as a ku80 homolog in citric acid-producing Aspergillus niger. Biosci Biotechnol Biochem 75:1594–1596
Huang X, Chen M, Lu X, Li Y, Li X, Li JJ (2014a) Direct production of itaconic acid from liquefied corn starch by genetically engineered Aspergillus terreus. Microb Cell Fact 13:108
Huang X, Lu X, Li Y, Li X, Li JJ (2014b) Improving itaconic acid production through genetic engineering of an industrial Aspergillus terreus strain. Microb Cell Fact 13:119
Huang X, Lu X, Li JJ (2014c) Cloning, characterization and application of a glyceraldehyde-3-phosphate dehydrogenase promoter from Aspergillus terreus. J Ind Microbiol Biotechnol 41:585–592
Ishibashi K, Suzuki K, Ando Y, Takakura C, Inoue H (2006) Nonhomologous chromosomal integration of foreign DNA is completely dependent on MUS-53 (human Lig4 homolog) in Neurospora. Proc Natl Acad Sci USA 103:14871–14876
Kanamasa S, Yamaoka K, Kawaguchi T, Sumitani J, Arai M (2003) Transformation of Aspergillus aculeatus using the drug resistance gene of Aspergillus oryzae and the pyrG gene of Aspergillus nidulans. Biosci Biotechnol Biochem 67:2661–2663
Klement T, Buchs J (2013) Itaconic acid–a biotechnological process in change. Bioresour Technol 135:422–431
Kopke K, Hoff B, Kuck U (2010) Application of the Saccharomyces cerevisiae FLP/FRT recombination system in filamentous fungi for marker recycling and construction of knockout strains devoid of heterologous genes. Appl Environ Microbiol 76:4664–4674
Krappmann S, Bayram O, Braus GH (2005) Deletion and allelic exchange of the Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryot Cell 4:1298–1307
Krappmann S, Sasse C, Braus GH (2006) Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end- joining-deficient genetic background. Eukaryot Cell 5:212–215
Lin YH, Li YF, Huang MC, Tsai YC (2004) Intracellular expression of Vitreoscilla hemoglobin in Aspergillus terreus to alleviate the effect of a short break in aeration during culture. Biotechnol Lett 26:1067–1072
Maruyama J, Kitamoto K (2008) Multiple gene disruptions by marker recycling with highly efficient gene-targeting background (∆ligD) in Aspergillus oryzae. Biotechnol Lett 30:1811–1817
Mattern IE, Unkles S, Kinghorn JR, Pouwels PH, van den Hondel CA (1987) Transformation of Aspergillus oryzae using the A. niger pyrG gene. Mol Gen Genet 210:460–461
Mizutani O, Kudo Y, Saito A, Matsuura T, Inoue H, Abe K, Gomi K (2008) A defect of LigD (human Lig4 homolog) for nonhomologous end joining significantly improves efficiency of gene-targeting in Aspergillus oryzae. Fungal Genet Biol 45:878–889
Mizutani O, Masaki K, Gomi K, Iefuji H (2012) Modified Cre-loxP recombination in Aspergillus oryzae by direct introduction of Cre recombinase for marker gene rescue. Appl Environ Microbiol 78:4126–4133
Slesiona S, Ibrahim-Granet O, Olias P, Brock M, Jacobsen ID (2012) Murine infection models for Aspergillus terreus pulmonary aspergillosis reveal long-term persistence of conidia and liver degeneration. J Infect Dis 205:1268–1277
Tani S, Tsuji A, Kunitake E, Sumitani J, Kawaguchi T (2013) Reversible impairment of the ku80 gene by a recyclable marker in Aspergillus aculeatus. AMB Express 3:4
Tevz G, Bencina M, Legisa M (2010) Enhancing itaconic acid production by Aspergillus terreus. Appl Microbiol Biotechnol 87:1657–1664
Wang BH, Xu Y, Li YP (2010) Use of the pyrG gene as a food-grade selection marker in Monascus. Biotechnol Lett 32:1631–1635
Acknowledgments
Thanks to Dr. Xiaoming Tan for valuable discussions. This work was supported by National Natural Sciences Foundation of China (Nos. 31400080 and 31500042), National High Technology Research and Development Program of China (Nos. 2015AA021003), the Key Research Program of the Chinese Academy of Sciences (No. KSZD-EW-Z-016).
Authors contributions
XL, XH and JL designed the experiments. XH, MC performed the experiments. XH, XL, JL and MC drafted the manuscript.
Supporting information
Supplementary Table 1—Primers used.
Supplementary Fig. 1—Construction of the plasmid pXH106
Supplementary Fig. 2—Characterization of the genotypes of At-∆ku80 mutant strain.
Supplementary Fig. 3—Genomic PCR analysis of the At-∆pyrG mutant strain.
Supplementary Fig. 4—Genomic PCR analysis of the At-loxP-pyrGAn mutants obtained in the complementation test of At-∆pyrG.
Supplementary Fig. 5—The DNA sequencing result of the PCR-amplified fragments from the pyrG An marker region of At-loxP-∆pyrGAn mutant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, X., Chen, M., Li, J. et al. Establishing an efficient gene-targeting system in an itaconic-acid producing Aspergillus terreus strain. Biotechnol Lett 38, 1603–1610 (2016). https://doi.org/10.1007/s10529-016-2143-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2143-y