
Abstract
High thermostability of enzymes is a prerequisite for their biotechnological applications. An organic solvent-tolerant and cold-active lipase, from the Stenotrophomonas maltophilia, was unstable above 40 °C in previous studies. To increase the enzyme stability, possible hydrogen-bond networks were simulated by the introduction of a salt bridge in a highly flexible region of the protein. Compared with the wild-type lipase, a mutant lipase (G165D and F73R) showed a >900-fold improvement in half-life at 50 °C, with the optimal activity-temperature increasing from 35 to 90 °C. Therefore, the hydrogen-bond strategy is a powerful approach for improving enzyme stability through the introduction of a salt bridge.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Enzymes that are stable under various conditions are a prerequisite for industrial applications in biotechnology and organic chemistry. However, many wild-type enzymes with poor stability are unsuitable in industrial application. Under high temperatures, many enzymes are easily inactivated because of heat-induced denaturation or structural damage (Teilum et al. 2011). For this reason, the development of enzyme variants with improved stability by protein engineering techniques will be helpful in further reduction of the cost of enzymes. Various strategies have been employed to improve enzyme stability, such as integrating salt bridges (Makhatadze et al. 2003; Gribenko et al. 2009), compacting the hydrophobic core (Pace et al. 2011), engineering disulfide bridges (Dürrschmidt et al. 2001), and stabilizing α-helices (Li et al. 2007; Facchiano et al. 1998).
Through atomic displacement parameters, named the B-factor, calculated by molecular dynamic (MD) simulations, several flexible regions responsible for initial denaturation of a lipase had been identified (Hespenheide et al. 2002; Gumulya and Reetz 2011; Reetz and Carballeira 2007; Reetz et al. 2006; Jochens et al. 2010). In the present study, experiments were designed to improve the stability of Stenotrophomonas maltophilia lipase by introducing a salt bridge in a flexible region, which leads to the reorganization and, thus, formation of a new hydrogen bond network. The resulting mutant lipase (G165D and F73R) displayed excellent stability at high temperature.
Materials and methods
Strains, plasmids, and site-directed mutagenesis
Escherichia coli BL21(DE3) and pET30a(+) were used as the expression host and vector, respectively. The Stenotrophomonas maltophilia lipase gene (NCBI accession No. KC014616) was available in the NCBI database. Site-directed mutagenesis was performed using the pET30a-Lipase plasmid as the template. The mutant lipase (G165D and F73R) was constructed using the QuikChange site-directed mutagenesis kit (Stratagene) and according to the instructions of the manufacturer. The sense and antisense primers used for mutagenesis are as follows: F73R mutation (sense primer 5′-CGCGTGGCcgcTGGGAATCCGGC-3′, antisense primer 5′-GATTCCCAgcgGCCACGCGAGCT-3′), and G165D mutation (sense primer 5′-TTGCGGCCgatCTGGTGACCGGA-3′, antisense primer 5′-GTCACCAGatcGGCCGCAACCAG-3′). The lower-case letters indicated mutagenic codons for the target amino acid residue. The plasmid containing the desired mutation was verified by DNA sequencing. The cloning, expression, purification and SDS-PAGE were carried out as previously described (Reetz et al. 2006).
Expression and purification of lipase
Recombinant E. coli BL21(DE3) was used for the expression of the lipase and was grown in 50 ml LB medium (50 μg kanamycin/ml) at 37 °C and 200 rpm. When the OD600 reached 0.8, IPTG was added at 0.3 mM, and the culture was incubated at 18 °C and 200 rpm for 12 h. Ni–NTA affinity chromatography was used to purify the His-tagged lipase on a 5-ml HisTrap FF column (GE Healthcare, USA) (Chong et al. 2009). To remove imidazole and NaCl, the protein was dialyzed against 50 mM Tris/HCl buffer (pH 7.5). The protein purity was tested on a 10 % (v/v) SDS-PAGE gel. Soluble protein content was estimated by the method of Bradford using bovine serum albumin as the standard.
Characterization of the mutant lipase stability
Hydrolysis of the pNP caproate was measured at 410 nm (Liu et al. 2006). In general, 20 μl of pNP caproate (20 mM) was added to 960 μl of Tris/HCl buffer (50 mM, pH 8.0). The reaction was initiated by adding 20 μl lipase at 60 °C. One unit of lipase activity was defined as the amount of enzyme needed to release 1 μmol pNP from pNP caproate per min at 60 °C. The thermostability of the lipase was determined by pre-incubation of the purified protein at 0–99 °C for 30 min. The residual activity was determined by measuring the pNP caproate as the substrate in 50 mM Tris/HCl buffer at 60 °C. The optimum temperature was assayed between 20 and 99 °C using pNP caproate as the substrate under standard conditions.
Homology modeling and molecular dynamics (MD) simulations
Homology model building was performed using the program SYBYL X-1.1 (Tripos, St Louis, MO, USA) by the fold-recognition method (Wu et al. 2011). For validation of the model, the stereochemical properties of the three-dimensional (3D) structure were analyzed by the Procheck program (Laskowski et al. 1993). All MD calculations were performed using the Amber11 software package with the Parm99 forcefield. The MD calculations for the salt bridge interactions and hydrogen bonds were analyzed by Discovery Studio 3.0. First, the lipase structure was given the CHARMM force field. The backbone of the protein was fixed. The protocol “Standard Dynamics Cascade” was employed to construct a protein structure of the mutant lipase, and then the structures were minimized. The RMSD was calculated for the lipase protein backbone atoms using the least-squares fit approach. The B-factors were calculated using the coordinates of the last 10 ns of the trajectories.
Results and discussion
Structural modeling of S. maltophilia lipase
The 3D model of the lipase was built using a bacterial esterase (Larsen et al. 2001) as the template model. The esterase (PDB: 1JU3) from Rhodococcus sp. MB1 showed the most sequence identity to the parent lipase (21 % identity). The 3D model of the lipase was built by the software SYBYL X-1.1 using the fold recognition method. The Z-score of the model was 67.3, indicating that the 3D model was “certain” (Z-score > 6.0). The calculated Ramachandran plot analysis revealed that more than 98 % of the residues in the model are in the favored and allowed regions, suggesting that the quality of the model was good. Analysis of the 3D model suggested the presence of a catalytic triad S115, D225, and H253. Each catalytic triad residue was mutated to Ala by site-directed mutagenesis, and the resultant mutations showed no activity in the presence of the pNP caproate substrate.
Structural basis of the regional flexibility and salt bridge
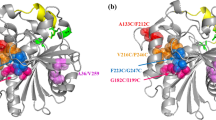
Based on the 3D model of the lipase, MD simulations with a heat stress of 323 K were carried out, obtaining the B-factor as shown in Fig. 1. Among several highly flexible regions in the model lipase, the sequences 160–200 was selected as the target in which lipase stability may be improved by incorporation of a salt bridge. This region contains several α-helix structures adjacent to the catalytic center (Fig. 2a). Therefore, a salt-bridge (G165D and F73R) was designed to stabilize the flexible region. The mutant lipase showed a new salt bridge compared with the wild-type enzyme (Fig. 2b). The obvious difference between the B-factor and RMSD results suggested that the mutation displayed lower flexibility (Figs. 1 and 3). The MD simulations revealed that six newly formed hydrogen bonds (201R–149S, 206S–132K, 352T–429N, 369N–252D, 397R–298Q) also contributed to reducing the flexibility of the global structure.

B-factor superposition of wild-type (WT) and mutated lipases at 323 K. The red line indicates the mutated lipase (G165D and F73R). The black line represents the WT lipase

Stereoview of the salt-bridge interactions in S. maltophilia lipase before and after substitutions at G165D and F73R. a The α-helix structure (160–200 amino acids) is shown in yellow, and the catalytic residues are red sticks. b The WT and mutant lipases are indicated as yellow and red, respectively. The hydrogen bonds between 165D and 73R are indicated by the green dashed lines

RMSD backbone results of WT and mutant lipases. The WT and mutant lipases are shown by black and red data, respectively
Expression and purification of the lipase
The WT and mutated lipases were expressed in the pET-30a-BL21 system and purified by Ni–NTA affinity chromatography, respectively. The expressed tagged-lipase was examined on an SDS-PAGE, and the expression levels were nearly the same (Fig. 4). Recombinant proteins in both cases were expressed in soluble forms, suggesting that the mutant lipase could be characterized. The purified lipase sample was more than 97 % pure, as assessed by SDS-PAGE analysis.

SDS-PAGE gel showing the expression and purification of recombinant His-tagged WT and mutant lipases. Lanes 1 and 2, crude and purified WT lipase; lanes 3 and 4, crude and purified mutant lipase. The crude lipase samples were obtained by extracting the supernatant of the induced cells. The purified lipases were prepared using a Ni–NTA column
Stability of the mutant lipase
The mutant lipase showed remarkable stability when compared with the wild-type enzyme. The mutant lipase displayed more than 85 % residual activity after incubation at 99 °C for 30 min, while the WT lipase was inactive above 80 °C (Fig. 5a). Furthermore, the lipase activity of mutant protein increased as the temperature was increased up to 90 °C (Fig. 5b). As a result, the optimal temperature was shifted from 35 to 90 °C. These results suggest that the salt bridge (165D and 73R) and the resulting hydrogen bonds led to an increase in stability of the mutant lipase against thermal denaturation.

The thermostability and optimal temperature of the lipases before and after substitutions. The filled circle and open circle indicate the mutated and WT lipases, respectively. a The remaining lipase activity was determined at 60 °C in 50 mM Tris/HCl buffer (pH 8.0) using pNP caproate as a substrate after 30 min incubation at different temperatures. b The enzymatic activity of the lipases was determined at various temperatures in 50 mM Tris/HCl buffer (pH 8.0) using pNP caproate as a substrate. The amount of lipase added was 1 mg/ml. The activities of the wild-type and mutated lipase were 55.8 and 11.3 U/ml, respectively
We incubated the wild-type and mutant at 90 °C for a much longer period of time (8 h) and the data are shown in the Fig. 6. The result showed that the mutated lipase was much more stable at 90 °C. Disappointingly, the hydrolytic activity of the mutant lipase decreased to about 20 % of that of the wild lipase. The specific activity of the wild lipase was 55.8 U/mg, and that of the mutant lipase was only 11.3 U/mg. In the analysis of enhanced stability of the mutant, we have identified pronounced long-range cooperative interactions (hydrogen bonds) caused by the formation of a salt bridge in the flexible region of the lipase (Wang et al. 1999).

The thermostability of the lipases before and after substitutions under 90 °C. The filled circle and open circle indicate the mutated and WT lipases, respectively. The remaining lipase activity was determined at 60 °C in 50 mM Tris/HCl buffer (pH 8.0) using pNP caproate as a substrate after different incubation time. The amount of lipase added was 1 mg/ml. The activities of the wild-type mutated lipase were 55.8 and 11.3 U/ml, respectively
References
Dürrschmidt P, Mansfeld J, Ulbrich-Hofmann R (2001) Differentiation between conformational and autoproteolytic stability of the neutral protease from Bacillus stearothermophilus containing an engineered disulfide bond. Eur J Biochem 268:3612–3618
Facchiano AM, Colonna G, Ragone R (1998) Helix stabilizing factors and stabilization of thermophilic proteins: an X-ray based study. Prot Eng 11:753–760
Gribenko AV, Patel MM, Liu J, McCallum SA, Wang C, Makhatadze GI (2009) Rational stabilization of enzymes by computational redesign of surface charge–charge interactions. Proc Nat Acad Sci 106:2601–2606
Gumulya Y, Reetz MT (2011) Enhancing the thermal robustness of an enzyme by directed evolution: least favorable starting points and inferior mutants can map superior evolutionary pathways. ChemBioChem 12:2502–2510
Hespenheide BM, Rader AJ, Thorpe MF, Kuhn LA (2002) Identifying protein folding cores from the evolution of flexible regions during unfolding. J Mol Graphics Model 21:195–207
Jochens H, Aerts D, Bornscheuer UT (2010) Thermostabilization of an esterase by alignment-guided focused directed evolution. Protein Eng Des Sel 23:903–909
Li J, Matsumura Y, Shinjo M, Kojima M, Kihara H (2007) A stable α-helix-rich intermediate is formed by a single mutation of the β-sheet protein, src SH3, at pH 3. J Mol Biol 372:747–755
Makhatadze GI, Loladze VV, Ermolenko DN, Chen X, Thomas ST (2003) Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J Mol Biol 327:1135–1148
Pace CN, Fu H, Fryar KL, Landua J, Trevino SR et al (2011) Contribution of hydrophobic interactions to protein stability. J Mol Biol 408:514–528
Reetz MT, Carballeira JD (2007) Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc 2:891–903
Reetz MT, Carballeira JD, Vogel A (2006) Iterative saturation muta-genesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed 45:7745–7751
Teilum K, Olsen JG, Kragelund BB (2011) Protein stability, flexibility and function. Biochim Biophys Acta 1814:969–976
Wang L, Duan Y, Shortle R, Imperiali B, Kollman PA (1999) Study of the stability and unfolding mechanism of BBA1 by molecular dynamics simulations at different temperatures. Prot Sci 8:1292–1304
Acknowledgments
The authors thank the National Basic Research Program of China (973 Program, No.2011CB710800), Hi-Tech Research and Development Program of China (863 Program, 2011AA02A209), National Natural Science Foundation of China (No. 20936002), and the Zhejiang University Foundation (No. 2013QNA4034) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jian-Ping Wu and Mu Li have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wu, JP., Li, M., Zhou, Y. et al. Introducing a salt bridge into the lipase of Stenotrophomonas maltophilia results in a very large increase in thermal stability. Biotechnol Lett 37, 403–407 (2015). https://doi.org/10.1007/s10529-014-1683-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-014-1683-2