
Abstract
Arterial media calcification is related to mitochondrial dysfunction. Protective mitophagy delays the progression of vascular calcification. We previously reported that lactate accelerates osteoblastic phenotype transition of VSMC through BNIP3-mediated mitophagy suppression. In this study, we investigated the specific links between lactate, mitochondrial homeostasis, and vascular calcification. Ex vivo, alizarin S red and von Kossa staining in addition to measurement of calcium content, RUNX2, and BMP-2 protein levels revealed that lactate accelerated arterial media calcification. We demonstrated that lactate induced mitochondrial fission and apoptosis in aortas, whereas mitophagy was suppressed. In VSMCs, lactate increased NR4A1 expression, leading to activation of DNA-PKcs and p53. Lactate induced Drp1 migration to the mitochondria and enhanced mitochondrial fission through NR4A1. Western blot analysis of LC3-II and p62 and mRFP-GFP-LC3 adenovirus detection showed that NR4A1 knockdown was involved in enhanced autophagy flux. Furthermore, NR4A1 inhibited BNIP3-related mitophagy, which was confirmed by TOMM20 and BNIP3 protein levels, and LC3-II co-localization with TOMM20. The excessive fission and deficient mitophagy damaged mitochondrial structure and impaired respiratory function, determined by mPTP opening rate, mitochondrial membrane potential, mitochondrial morphology under TEM, ATP production, and OCR, which was reversed by NR4A1 silencing. Mechanistically, lactate enhanced fission but halted mitophagy via activation of the NR4A1/DNA-PKcs/p53 pathway, evoking apoptosis, finally accelerating osteoblastic phenotype transition of VSMC and calcium deposition. This study suggests that the NR4A1/DNA-PKcs/p53 pathway is involved in the mechanism by which lactate accelerates vascular calcification, partly through excessive Drp-mediated mitochondrial fission and BNIP3-related mitophagy deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The global incidence of diabetes mellitus (DM) is still growing steadily. Diabetic cardiovascular complications are the leading cause of high morbidity and mortality in diabetics [1, 2]. Vascular calcification, a common vascular complication in patients with diabetes, has brought difficulties to patients with coronary artery disease and chronic kidney disease [3, 4]. Inflammation, oxidative stress, and apoptosis, caused by phosphate deposits or advanced glycation end-products, play an important role in calcium deposition and osteoblastic phenotype transition of vascular smooth muscle cells (VSMCs), the main cell type of vascular media [5,6,7,8]. Damaged mitochondria are the main sources of reactive oxygen species (ROS) generation, and oxidative stress itself together with cytochrome-c (cyt-c) leakage initiates apoptosis [9, 10]. Therefore, the removal of damaged mitochondria is required to prevent vascular calcification.
The roles of mitophagy and mitochondrial fission in regulating mitochondrial homeostasis have been extensively studied [11]. Previous studies showed that excessive mitochondrial fission induces ROS overproduction under stress conditions [12]. Uncontrolled mitochondrial fission produces large amounts of mitochondrial debris that damage the mitochondrial genome and disrupt normal mitochondrial respiration and ATP production [13]. In addition, abnormal fission promotes mitochondrial permeability transition pore (mPTP) opening and cyt-c leakage, eventually inducing mitochondrial-dependent apoptosis [14, 15]. Dynamin-related protein 1 (Drp1), a key molecule involved in mitochondrial fission, exacerbates cardiovascular calcification in an oxidative stress-dependent manner [16]. Mitophagy effectively removes damaged mitochondria and avoids excessive ROS generation [17]. Interestingly, we found that mitochondrial biogenesis also depends on mitophagy, so that the quantity and quality of mitochondria are relatively balanced, finally alleviating VSMC calcification [18]. Therefore, mitophagy and mitochondrial fission are both implicated in the process of vascular calcification.
It has recently been shown that mitochondrial fission and mitophagy are regulated by p53, which assists in the transfer of Drp1 from the cytoplasm to the mitochondria to promote the onset of fission [19, 20]. At the same time, phosphorylated p53 effectively inhibits the expression of mitophagy receptor BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) [21]. p53 is phosphorylated by DNA-dependent protein kinase catalytic subunit (DNA-PKcs) [22], an important enzyme participating in the repair of DNA double-strand breaks [23]. Under normal circumstances, DNA-PKcs binds to Ku80, thus providing a mechanism for internal repair of cells. However, in response to irreversible injury, DNA-PKcs interacts with and phosphorylates p53 at Ser15 to activate p53-dependent apoptosis [24, 25]. NR4A1 is a nuclear receptor and is involved in atherosclerosis, although there is controversy about its effects on atherosclerosis [26, 27]. High levels of NR4A1 block the binding of DNA-PKcs to Ku80 and enhance binding of DNA-PKcs to p53 [28]. Besides, NR4A1 is highly expressed in VSMC and is a sensor of oxidative stress [29]. Furthermore, NR4A1 serves as a damaging factor in cardiac microvascular ischemia reperfusion injury through elevation of fission and suppression of mitophagy [30]. Whether this signaling pathway (NR4A1/DNA-PKcs/p53) could affect vascular calcification at the VSMC level by regulating mitochondrial fission and mitophagy remains unknown.
Atherosclerotic disease is closely related to metabolic reprogramming [31, 32], and lactate, by-product of anaerobic glycolysis, has been reported to be associated with cardiovascular disease. Clinical studies have demonstrated that patients with diabetes are usually accompanied by elevated levels of blood lactate [33], which also correlate with carotid atherosclerosis and hypertension [34, 35]. Vascular calcification is highly similar to atherosclerosis, and VSMC synthetic transformation is a common feature [36]. Furthermore, lactate is reported to promote the synthetic phenotype transition in VSMC [37]. These reports suggest that lactate may be linked with vascular calcification. We previously demonstrated that lactate accelerates VSMC calcification through suppression of BNIP3-mediated mitophagy [38],however, whether lactate regulates mitochondrial fission while regulating mitophagy, and the potential involvement of the NR4A1-mediated pathway remain to be clarified.
In the present study, we focused on three issues: (1) whether lactate promotes vascular calcification in calcified aorta models; (2) whether mitochondrial fission and mitophagy participate in the process by which lactate accelerates vascular calcification; and (3) whether lactate disrupts mitochondrial homeostasis via the NR4A1/DNA-PKca/p53 pathway in VSMCs.
Materials and methods
Calcification model
All experiments in vivo were approved by the Animal Care and Research Committee of Southeast University, and all animal procedures were performed in accordance with the Guidelines of Animal Experiments from the Committee of Medical Ethics, the National Health Department of China (1998).
Briefly, 6-week old male Wistar rats (160–180 g, Experimental Animal Center in Shanghai, China) were divided into 4 groups. The first group was the control group, age-matched rats were only given a standard rat chow diet (60% carbohydrates, 22% protein, 10% fat, 8% fiber and other ingredients) for 9 weeks (n = 6); The second group was the vitamin D3 + nicotine (VDN) group, rats were fed with a standard chow diet for 9 weeks and were administered with vitamin D3 (300 000 U/kg, intramuscularly, Sigma-Aldrich, St. Louis, MO, USA) and nicotine (25 mg/kg, 5 ml/kg, PO, Sigma-Aldrich) to induce arterial media calcification (n = 12). Nicotine administration was repeated after 12 h on the same day. The remaining two groups (DM and DM + VDN) of rats are all DM background. Six-week old rats were fed a high fat diet (HFD, 50% carbohydrates, 30% fat (20% lard and 10% soybean oil), 11% protein, 2.5% cholesterol, 6.5% fiber and other ingredients) (Animal Center, Health Science Center, Southeast University) for 8 weeks, followed by a single dose of streptozotocin (STZ, 25 mg/kg, intraperitoneally, Sigma-Aldrich) (n = 18). Animals were considered to be diabetic when the blood glucose levels were over 16.7 mM. All of the diabetic rats were maintained on a standard chow for 1 week after the start of the STZ administration. Two-thirds of the diabetic rats were then treated with VDN. All the rats were then allowed to recover for 2 weeks.
Organ culture of aorta
Thoracic aortas were dissected from Wistar rats and cultured ex vivo according to previous protocols [39]. Half of the VDN or DM + VDN thoracic aortas were incubated in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium (DMEM) and Ham's F12 medium with 15% fetal bovine serum (FBS) and lactate (10 mM) for 2 weeks. The remaining groups were cultured with complete medium (DMEM + F12 + 15% FBS) for 2 weeks. The medium was renewed twice a week. Please see the figure blow.

Group treatment protocols ex vivo Open bars represent a standard chow diet, gray bars represent a high fat diet, dark blue bars represent a lactate treatment, and green bars represent a complete medium treatment. The vertical dashed lines represent the injection of STZ. The vertical solid lines represent the vitamin D3 and nicotine treatment.
Cell culture
Primary VSMCs were isolated from 6-week-old Sprague Dawley rat thoracic aortas (60–80 g, Experimental Animal Center in Shanghai, China) according to previous protocols. VSMCs between passages 4 and 7 were incubated in a 1:1 mixture of DMEM and Ham's F12 medium with 10% FBS and antibiotics at 37 °C with 5% CO2. VSMC calcification was induced with β-glycerophosphate (β-GP, Sigma-Aldrich) according to previous protocols [5].
Measurement of the calcium content and ALP activity
Calcium content of calcified thoracic aortas or VSMCs was measured using the Calcium Assay kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) and normalized to the total protein content with a Bicinchoninic Acid (BCA) Protein Assay kit (KeyGEN Biotechnology, Jiangsu, China).
For ALP activity measurement, the calcified VSMCs were solubilized with RIPA lysis buffer (Beyotime Biotechnology). After centrifugation, the supernatants were examined with the ALP activity kit (Nanjing Jiancheng Bioengineering Institute) and normalized to the total protein content.
Alizarin red S staining
The calcified VSMCs were fixed in 4% paraformaldehyde for 30 min at room temperature, washed twice with phosphate buffer saline (PBS), and then stained with 2% Alizarin red S (pH 8.4) (ScienCell Research Laboratories, Shanghai, China) for another 30 min at 37 °C. Excess reagent was removed by washing with PBS. The calcium nodules were observed under a microscope.
Aortic segments were fixed in 4% paraformaldehyde and embedded in paraffin. Aortic samples were cut into 6 μm in thickness. Sections were deparaffinized, stained with 2% alizarin red for 5 min and washed with PBS. And then the sections were soaked in anhydrous acetone solution for 30 s, mixed solution of anhydrous acetone and xylene (volume ratio = 1:1) for 15 s and anhydrous xylene for 1 min. The calcium phosphate salts were visualized as a red staining.
von Kossa staining
Paraffin sections from aortas were dewaxed and hydrated. The slices were immersed in 1% silver nitrate for 30 min under an intense sunbeam, and were then washed 3 times with deionized water. Subsequently, 5% sodium thiosulfate (Sigma-Aldrich) was added for 5 min to remove un-reacted reagent. The calcium phosphate salts were visualized as a black staining.
Mitochondrial function analysis
mPTP opening was measured based on a mPTP colorimetric detection kit (Genmed Scientifics Inc., Shanghai, China) according to the relative fluorescence units (RFUs) of mitochondrial volume changes, which were determined by a spectrophotometer (MULTISKAN GO, Thermo Fisher Scientific, USA).
Mitochondrial membrane potential was evaluated via JC-1 staining (KeyGEN Biotechnology) by flow cytometry (BD Biosciences, USA). Briefly, after treatment, VSMCs were washed twice with PBS and then stained with JC-1 probes for 20 min at 37 °C. VSMCs with healthy mitochondria were distributed in Q2. While VSMCs with mitochondrial membrane potential decline were distributed in Q3.
The adenosine triphosphate (ATP) levels in the VSMCs were detected using an ATP assay kit (Beyotime Biotechnology) according to the protocol. Briefly, 20 μl of sample or standard were added to the test well or tube and mixed quickly with a micropipette at least 2 s. Luminescence was measured using a microplate reader (Bio-Rad, Shanghai, China).
For oxygen consumption rate (OCR) determination, VSMCs were seeded into Seahorse 24-well plates at 8000 cells/well and were then treated as previously reported [40]. OCR was measured by a Seahorse XFe24 analyzer (Seahorse Bioscience, Boston, MA, USA). A Mito-stress kit (Seahorse Bioscience), oligomycin, carbonylcyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and rotenone were added according to the manufacturer’s instructions. All results were normalized to the cell number. Basal respiration = (last rate measurement before first injection)—(minimum rate measurement after antimycin injection). Maximal respiration = (maximum rate measurement after FCCP injection) − (minimum rate measurement after antimycin injection).
Transfection
To silence NR4A1, DNA-PKcs and p53, each of three small interfering RNAs (siRNAs) were designed (Shanghai Genechem Co., Ltd.). The siRNAs were transfected into cells with Lipofectamine 2000 (Invitrogen Life Science, Grand Island, NY) for 24 h according to the manufacturer’s protocol. The transfection efficiency was examined by western blotting.
To detect the autophagic flux, VSMCs were transfected with mRFP-GFP-LC3 adenovirus (Shanghai Hanbio Biotechnology Co., Ltd.) for 4 h (MOI = 5). After transfection, the medium was renewed with complete medium for 12 h and then given interventions. At 48 h after transfection, autophagosomes (yellow dots) and autolysosomes (red dots) were detected under a confocal microscope (FV10i, Olympus, Japan).
Mitochondria morphology observation
The VSMCs were washed twice with PBS, and then cultured with red mito-tracker (Genmed Scientifics Inc.) for 20 min. Excess reagent was removed by washing with PBS. The mitochondria morphology was observed under a confocal microscope.
Mitochondrial isolation
Mitochondrial and cytosolic fractions were obtained using a mitochondrial isolation kit (Beyotime Biotechnology). Briefly, after the VSMCs were incubated in ice-cold mitochondrial lysis buffer for 15 min, the cell suspension was homogenized for 20 strokes. Next, the homogenate was centrifuged at 1000×g for 10 min at 4 °C to remove the nuclei and unbroken cells. The supernatant was then collected and centrifuged again at 11,000×g for 10 min at 4 °C to obtain the mitochondrial fraction. For western blotting analysis, the mitochondrial fractions were stored in mitochondrial lysis buffer containing PMSF.
Western blotting analysis
The aortas or VSMCs were lysed according to the manufacturer’s instructions, and the protein concentration was measured using the BCA Protein Assay kit. Antibodies against microtubule-associated protein 1 light chain 3 (LC3) (CST4108) (1:2000), SQSTM1 (p62) (CST23214) (1:500), alpha-SM-actin (α-SMA) (CST19245) (1:1000), Cleaved-Caspase3 (CST9661) (1:1000), Bcl-2 (CST3498) (1:1000), and Bax (CST14796) (1:1000) were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-runt-related transcription factor 2 (RUNX2) (ab76956) (1:1000), anti-bone morphogenetic protein 2 (BMP-2) (ab214821) (1:1000), anti-Drp1 (ab184247) (1:1000), anti-mitochondrial fission factor (Mff) (ab81127) (1:1000), anti-BNIP3 (ab109362) (1:1500), anti-NR4A1 (ab109180) (1:1000), anti-DNA-PKcs (ab70250) (1:2000), anti-p53 (Ser15) (ab1431) (1:1000), and anti-translocase of outer mitochondrial membrane 20 (TOMM20) (ab186734) (1:500) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-translocase of outer mitochondrial membrane 40 (TOMM40) (66,658–1-Ig) (1:500), anti-voltage-dependent anion channel 1 (VDAC1) (55,259–1-AP) (1:1000), and anti-optic atrophy 1 (OPA1) (27,733–1-AP) (1:1000) were obtained from Proteintech (Wuhan, China). Anti-β-actin (1:3000) and all secondary antibodies (1:5000) were provided by Biosharp (Anhui, China). Subsequently, 20–40 μg of protein was loaded onto a SDS-PAGE gel and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin (BSA) for 1.5 h. The membranes were incubated with different primary antibodies overnight at 4 °C and then visualized using anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase for 1 h at room temperature. The blots were detected using electrochemiluminescence (ECL), and the results were quantified with the Image-Pro Plus 6.0 software.
Immunofluorescence staining
The VSMCs were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 20 min, and then blocked with 5% BSA for 0.5 h at room temperature. The cells were incubated with anti-LC3 (1:500) and anti-TOMM20 (1:250) antibodies at 4 °C overnight, followed by the appropriate secondary antibodies for 0.5 h in the dark. The images were visualized using a confocal microscope, and the results were quantified with the Image-J software.
Immunohistochemistry
Briefly, after dewaxing and hydration of sections, tissue slides were treated with 3% H2O2 for 10 min, then were incubated with the primary antibodies against α-SMA (1:500), RUNX2 (1:500), Mff (1:200), and BNIP3 (1:150) overnight at 4 °C, followed by incubation with the secondary antibody conjugated to horseradish peroxidase. The antibodies were detected as a brown color. The tissue sections were scored according to the degree of staining (0–3 divided into negative staining, light yellow, light brown, dark brown) and positive range under light microscope (1–4 points were 0–25%, 26–50%, 51–75%, 76–100%), then the final score can be carried out the sum.
Transmission electron microscopy (TEM)
The VSMCs were fixed in 2.5% glutaraldehyde (electron microscopy grade) at 4 °C for 2 h, dehydrated in an ethanol series, and embedded in Epon resin. Representative areas were chosen for ultrathin sections and viewed with a Hitachi TEM system at an accelerating voltage of 80 kV. Digital images were obtained by an AMT imaging system (Advanced Microscopy Techniques Co., Danvers, MA, USA). For mitochondrial morphology and structural changes observations. At least 5–7 cells per condition were imaged.
Statistical analysis
All of the experiments were independently repeated at least three times. All data are presented as the mean ± standard deviation (SD). Statistical analyses were performed using Statistical Package for Social Science (SPSS) 22.0 (SPSS, Chicago, IL, USA). Two-tailed Student’s t-tests and one- or two-way ANOVA with post hoc comparisons by Tukey’s multiple comparisons test were used to compare the results. Values of P < 0.05 were considered significant.
Results
Lactate accelerated vascular calcification and caused apoptosis ex vivo
We first investigated the effects of lactate on vascular calcification in Wistar rats. As described in the methods section, we divided the aortas into six groups (con, VDN, VDN + lactate, DM, DM + VDN, and DM + VDN + lactate). Since the blood lactate levels of diabetic rats were about 5 mM, while the blood lactate levels of diabetic calcification rats were about 7 mM in our study (data not shown), so we chose a lactate concentration of 10 mM for ex vivo culture. As shown in Fig. 1a, alizarin S red and von Kossa staining revealed a degree of calcification in the VDN group in both the diabetic and non-diabetic models, while lactate significantly expanded and enhanced the area and degree of the vascular media calcification. Determination in the calcium content in the thoracic aorta was consistent with the results observed in Fig. 1a, and lactate further elevated calcium content based on VDN modeling (Fig. 1b). The phenotypic transition of VSMC in the vascular media is important for calcification. We analyzed the expression of RUNX2 and BMP-2, which are key nodes in VSMC osteoblastic differentiation pathway [41], in thoracic aortas by immunohistochemistry and western blotting. As shown in Fig. 1c, d, lactate caused the VSMC contraction phenotype to be lost and differentiation of an osteoblastic phenotype.

Lactate accelerated vascular calcification ex vivo. a Calcium nodules were stained with alizarin S red (red staining) or von Kossa staining (black staining). Scale bar, 100 μm b calcium content of thoracic aortas was detected. c Immunohistochemical analysis of thoracic aorta α-SMA and RUNX2. Scale bar, 50 μm d Western blot analysis of NR4A1, osteoblastic phenotype transition-related molecules (α-SMA, RUNX2, BMP-2), and apoptosis-related molecules (Cleaved-Caspase3, Bax, Bcl-2). *P < 0.05 compared with the con group. #P < 0.05 compared with the VDN group. &P < 0.05 compared with the DM group. @P < 0.05 compared with the DM + VDN group. Data are presented as the mean ± standard deviation of three experiments (Color figure online)
Since apoptosis is inducible for the osteoblastic phenotype transition of VSMC and calcium nodule formation. Furthermore, we previously reported that lactate treatment leads to VSMC apoptosis [38]. Therefore, we evaluated apoptosis-related protein levels in the thoracic aortas. The expression Cleaved-Caspase3 and Bax was markedly elevated in the VDN + lactate groups compared with that in the VDN groups, whereas the expression of anti-apoptotic Bcl-2 was reduced, which demonstrated a pro-apoptotic role of lactate (Fig. 1d). These results confirmed the pro-calcification effects of lactate ex vivo, and further linked lactate, apoptosis and vascular calcification.
Lactate regulated mitochondrial fission and mitophagy ex vivo
Mitochondrial homeostasis is closely related to apoptosis, while the former relies mainly on mitochondrial structural stability and effective clearance of abnormal mitochondria. We detected changes in autophagy-related molecules and mitochondrial membrane proteins by immunohistochemistry and western blotting. In calcified thoracic aortas, the overall autophagy levels and BNIP3 expression increased significantly, whereas the mitochondrial membrane protein levels decreased, suggesting an intracellular self-regulation. As expected, subsequent lactate intervention reversed these results (Fig. 2).

Lactate regulated mitochondrial homeostasis ex vivo. a Immunohistochemical analysis of thoracic aorta Mff and BNIP3. Scale bar, 50 μm b Western blot analysis of mitophagy-related molecules (LC3-II, p62, BNIP3), mitochondrial membrane proteins (TOMM20 and TOMM40), and mitochondrial fission proteins (mito-Drp1, cyto-Drp1, Mff, OPA1). *P < 0.05 compared with the con group. #P < 0.05 compared with the VDN group. &P < 0.05 compared with the DM group. @P < 0.05 compared with the DM + VDN group. Data are presented as the mean ± standard deviation of three experiments
Previously, it has been reported that Drp1, which is a key molecule involved in mitochondrial fission, accelerates vascular calcification by activating oxidative stress [16],we also found that the protein levels of the Drp1 receptor, Mff, were increased in the calcified thoracic aortas, and the levels of OPA1, a key molecule involved in mitochondrial fusion, were distinctly suppressed against both the diabetic and non-diabetic background. Compared with the non-calcified group, mito-Drp1 expression was significantly elevated in the calcified group, and cyto-Drp1 protein levels were reduced, suggesting Drp1 migration. Lactate exacerbated the migration of cyto-Drp1 on the basis of calcification (Fig. 2). Overall, these results indicated that lactate regulates both mitochondrial fission and mitophagy to disturb mitochondrial homeostasis.
Lactate accelerated calcification in VSMCs through NR4A1 activation
To demonstrate the role of NR4A1 in vascular calcification, we measured NR4A1 protein levels in calcified thoracic aortas. Compared with non-calcified tissues, the VDN groups had higher NR4A1 protein levels that were exacerbated in the VDN + lactate groups (Fig. 1d). To further confirm the pro-calcification role of NR4A1 in vitro, we silenced NR4A1 in VSMCs by transfection with three siRNA sequences (si-NR4A1-1-3), and the transfection efficiencies were determined by western blotting. We selected si-NR4A1-3 sequence against NR4A1 (approximately 80% silencing efficiency) for the subsequent experiments (Supplementary Fig. 1A).
VSMCs were divided into the following groups: control, β-GP, β-GP + lactate (10 mM), β-GP + lactate (10 mM) + si-NR4A1, β-GP + lactate (10 mM) + scrambled siRNA, and β-GP + lactate (10 mM) + cytosporone B (CsnB) (10 μg/ml) (NR4A1 inducer). The concentration of lactate used here was borrowed from previous report [37]. Compared with the lactate untreated groups, NR4A1 expression was significantly upregulated in the calcified + lactate VSMC models after 24 h of lactate treatment. Lactate increased RUNX2 and BMP-2 protein expression and decreased α-SMA levels in calcified VSMCs compared with the levels detected in the β-GP alone group. These effects were reversed by siRNA-mediated NR4A1 knockdown. In contrast, CsnB-induced NR4A1 overexpression enhanced the osteoblastic phenotype transition of VSMC (Fig. 3a). In addition, lactate treatment increased ALP activity and the calcium deposition content after 7 days through NR4A1 activation (Fig. 3b, c). Calcified nodule formation was also significant after 21 days in the β-GP + lactate group (Fig. 3d), and persistent CsnB intervention significantly increased the number of calcium nodules. All these results revealed that NR4A1 plays an important role in lactate-accelerated VSMC calcification.

Lactate accelerated osteoblastic phenotype transition of VSMC and calcium deposition through NR4A1. Normal or calcified VSMCs were divided into six groups: con, β-GP, β-GP + lactate (10 mM), β-GP + lactate (10 mM) + si-NR4A1, β-GP + lactate (10 mM) + scrambled siRNA, and β-GP + lactate (10 mM) + CsnB (10 μg/ml). Each group was incubated for 24 h. a Western blot analysis of NR4A1 and osteoblastic phenotype transition-related molecules (α-SMA, RUNX2, BMP-2). b, c ALP activity and calcium deposition were detected after 7 days intervention. d After 21 days treatment, calcium nodule formation was visualized by alizarin red S staining. At least 3–5 images per condition were imaged. Scale bar, 50 μm. *P < 0.05 compared with the con group. #P < 0.05 compared with the β-GP group. &P < 0.05 compared with the scrambled siRNA group. @P < 0.05 compared with the β-GP + lactate group. Data are presented as the mean ± standard deviation of three experiments
Lactate activated Drp1-related mitochondrial fission through NR4A1 upregulation
Since excessive mitochondrial fission is implicated in mitochondrial dysfunction, and Drp1 is involved in vascular calcification [16], we then investigated the relationship between lactate, NR4A1, and Drp1-mediated fission. We first observed the morphological changes of mitochondria under lactate treatment. The mitochondrial length was examined to quantify mitochondrial fission. In normal control VSMCs, the mitochondria were cable-like, thin strips. In calcified cells, some of the mitochondria were divided into several fragments, and this damage to the mitochondrial structure was exacerbated by the addition of lactate. However, mitochondria in the NR4A1-knockout cells showed a predominantly elongated form despite lactate treatment. CsnB-induced activation of NR4A1 aggravated the damaging effects of lactate on mitochondrial fission in the calcified environment (Fig. 4a).

Lactate aggravated Drp1-mediated mitochondrial fission through NR4A1. a VSMCs were stained with mito-tracker red to evaluate the mitochondrial fragmentation. The length of mitochondria was measured. At least 8–10 cells per condition were imaged. Scale bar, 5 μm. b, c Change of Drp1 cellular distribution. Mff and OPA1 protein levels were determined by western blotting. *P < 0.05 compared with the con group. #P < 0.05 compared with the β-GP group. &P < 0.05 compared with the scrambled siRNA group. @P < 0.05 compared with the β-GP + lactate group. Data are presented as the mean ± standard deviation of three experiments
Migration of cyto-Drp1 to the mitochondria is essential for mitochondrial fission [42]. As shown in Fig. 4b, c, lactate increased mito-Drp1 and Mff expression but reduced cyto-Drp1 and OPA1 content in the calcified VSMCs compared with the non-lactate groups. These tendencies were reversed by NR4A1 block. Notably, CsnB-induced NR4A1 overexpression exacerbated the inducible effects of lactate on Drp1 migration. Altogether, these results indicated that lactate activates Drp1-related mitochondrial fission through NR4A1.
NR4A1 suppressed the autophagic flux and BNIP3-mediated mitophagy
Mitophagy is another important mechanism responsible for maintaining mitochondrial homeostasis. We first evaluated the role of NR4A1 in lactate-inhibited autophagic flux. After lactate treatment for 24 h, the LC3-II protein level was decreased and p62 was increased compared with the levels detected in the β-GP group. The trend of LC3-II and p62 expression was reversed after NR4A1 knockdown; however, CsnB pre-treatment enhanced the trend (Fig. 5a). To further determine alterations in the autophagic flux, the VSMCs were transfected with the mRFP-GFP-LC3 adenovirus for confocal detection. The autophagy inducer rapamycin (Rapa) (10 μM, Sigma-Aldrich) was used as a positive control, and chloroquine (CQ) (10 μM, Sigma-Aldrich) was used to block fusion of the autophagosome and lysosome. As shown in Fig. 5b, Rapa treatment increased the numbers of both GFP (green) and mRFP (red) labeled cells and a large number of red (autolysosomes) cells were observed. There was no significant difference in the autolysosome numbers between the CQ group and the β-GP + lactate group, indicating that lactate blocked the subsequent processes in a large number of autophagosomes. Then, we found that red/yellow (autolysosome/autophagosome) dots ratio was markedly decreased in the VSMCs cultured with calcium medium and lactate compared with the calcified VSMCs. However, after NR4A1 deletion, the autolysosome/autophagosome ratio was increased, suggesting that a part of the autophagosomes were degraded by lysosomes. As expected, CsnB inhibited the combination of lysosomes and autophagosomes. We could conclude that NR4A1 was involved in the process of lactate blocking autophagic flux.


Lactate blocked autophagic flux and suppressed BNIP3-related mitophagy via NR4A1 up-regulation. a After 24 h treatment, LC3-II, p62, TOMM20, and BNIP3 protein levels were evaluated by western blotting. b VSMCs were pre-treated with Rapa (10 μM) for 4 h or CQ (10 μM) for 12 h. Then VSMCs were transfected with mRFP-GFP-LC3 adenovirus for 4 h for fluorescent analysis. The yellow puncta indicate autophagosome, and the free red puncta indicate autolysosome. Scale bar, 10 μm. At least 8–10 cells per condition were imaged. Quantification represents the ratio of autolysosome/autophagosome in each group. c Immunofluorescent images double labelled by LC3-II (green) and TOMM20 (red). At least 8–10 cells per condition were imaged. Scale bar, 5 μm. *P < 0.05 compared with the con group. #P < 0.05 compared with the β-GP group. &P < 0.05 compared with the scrambled siRNA group. @P < 0.05 compared with the β-GP + lactate group. Data are presented as the mean ± standard deviation of three experiments (Color figure online)
Next, we investigated the effects of NR4A1 regulation on mitophagy. The lower levels of TOMM20 and TOMM40 (translocases of the outer mitochondrial membrane) and elevated BNIP3 protein levels induced by β-GP were reversed by incubation with lactate, indicating attenuated mitochondrial clearance. NR4A1 knockdown promoted mitochondrial clearance, whereas NR4A1 overexpression had the opposite effect (Fig. 5a). In addition, NR4A1 knockdown promoted autophagosome (LC3-II green) and mitochondria (TOMM20 red) fusion compared with that observed in the scrambled siRNA group, thereby restoring mitochondrial networks and suggesting mitophagy activation (Fig. 5c). Thus, it was demonstrated that NR4A1 participates in lactate-inhibited autophagic flux and BNIP3-mediated mitophagy.
NR4A1 deletion preserved mitochondrial structure and respiratory function in VSMCs
When cells are subjected to stress and injury, the mitochondria enlarge and the mPTPs open, releasing large amounts of cyt-c, which leads to a decline in the membrane potential decline and stimulation of the mitochondrial apoptotic pathway [43]. In our study, calcified VSMCs exhibited an increased rate of mPTP opening, and the effect was enhanced by lactate treatment,however, NR4A1 knockdown decreased the rate of mPTP opening, whereas CsnB pre-treatment promoted mPTP opening (Fig. 6a). TEM analysis was used to provide direct evidence of mitochondria swelling. As shown in Fig. 6b, when the cells were in the process of osteogenic differentiation, the mitochondria enlarged and its internal structure was significantly damaged. Lactate stimulation caused mitochondrial enlargement to such a degree that the internal ridge structure was almost completely destroyed. In the NR4A1 knockout group, the degree of mitochondrial swelling was significantly reduced, whereas NR4A1 overexpression exacerbated the destruction of the inner and outer mitochondrial membranes, leading to discharge of large amount of the contents. Further quantitative analysis showed that NR4A1 regulation had a significant effect on the number of damaged mitochondria, and that NR4A1 knockout could partially offset the destruction caused by lactate. JC-1 staining showed a larger percentage of mitochondria distributed in Q3 after lactate exposure than those in the control or β-GP groups; NR4A1 suppression reduced this percentage, suggesting an alleviated decline in the mitochondrial membrane potential (Fig. 6c). Western blotting analysis of Cleaved-Caspase3, Bax, and Bcl-2 expression revealed a pro-apoptotic role for NR4A1 (Fig. 6d). These results suggested that NR4A1 knockout has a protective effect on the integrity of the mitochondrial structure and cell apoptosis in the presence of lactate.


NR4A1 knockdown could preserve mitochondrial structure and respiratory function. a The RFU of mPTP opening was evaluated by spectrophotometry at 505 nm. b Mitochondrial morphology and structural changes under TEM. At least 5–7 cells per condition were imaged. Scale bar, 0.2 μm. c The mitochondrial membrane potential was measured via JC-1 staining with flow cytometry. d Western blot analysis of apoptosis-related molecules (Cleaved-Caspase3, Bax, Bcl-2). e Cellular ATP production determination. f The OCR assay was used to observe the mitochondrial respiratory function. *P < 0.05 compared with the con group. #P < 0.05 compared with the β-GP group. &P < 0.05 compared with the scrambled siRNA group. @P < 0.05 compared with the β-GP + lactate group. Data are presented as the mean ± standard deviation of three experiments
Mitochondria are the factories of cellular energy. The ATP is actually derived from the proton chemical gradient that forms the mitochondrial inner membrane potential. Since the decline in the mitochondrial membrane potential was related to NR4A1, as expected, NR4A1 overexpression further reduced ATP generation based on the role of lactate (Fig. 6e). We then measured OCR to evaluate mitochondrial respiratory function. The OCR was decreased after lactate treatment compared with that of the calcified VSMCs, and NR4A1 knockout restored the OCR. Moreover, CsnB treatment further lowered the OCR. Compared with the β-GP + lactate group, the basal and maximum respiration rates were both improved after NR4A1 silencing, both of which were decreased by pre-treatment with CsnB (Fig. 6f). We concluded that mitochondrial respiratory function is further affected by the destruction of mitochondrial structure.
The NR4A1/DNA-PKcs/p53 pathway participated in Drp1 and BNIP3 regulation
Finally, to explore the involvement of the NR4A1-related signaling pathway in mitochondrial fission and mitophagy regulation, DNA-PKcs was first silenced. After transfecting VSMCs with three siRNA sequences designed to mediate DNA-PKcs knockdown, we selected si-DNA-PKcs-2 sequence against DNA-PKcs (approximately 80% silencing efficiency) (Supplementary Fig. 1B). As shown in Fig. 7a, lactate intervention increased the protein levels of DNA-PKcs and p53(Ser15) compared with that in the non-lactate groups; however, the expression of DNA-PKcs and p53 phosphorylation was significantly reduced after NR4A1 knockdown, suggesting that the DNA-PKcs and p53 are downstream molecules of NR4A1. In addition, DNA-PKcs blocking ablated p53 phosphorylation. These findings demonstrated the involvement of the NR4A1/DNA-PKcs/p53 axis in lactate-treated calcified VSMCs.

NR4A1 activated p53-mediated mitochondrial fission and mitophagy through DNA-PKcs up-regulation. a DNA-PKcs and p53 expression was decreased after NR4A1 block. P53 was regulated by DNA-PKcs. b, c P53 knockdown alleviated mitochondrial fission, detected by Mff protein levels and mitochondria average length. At least 8–10 cells per condition were imaged. Scale bar, 5 μm. b, d P53 silencing promoted BNIP3-mediated mitophagy, evaluated by BNIP3 protein levels and LC3-II (green) and TOMM20 (red) co-localization. At least 8–10 cells per condition were imaged. Scale bar, 5 μm. *P < 0.05 compared with the con group. #P < 0.05 compared with the β-GP group. &P < 0.05 compared with the scrambled siRNA group. @P < 0.05 compared with the β-GP + lactate group. Data are presented as the mean ± standard deviation of three experiments (Color figure online)
Next, we performed siRNA-mediated silencing of p53 and used western blot analysis to confirm that, of the three candidate siRNAs, the highest silencing efficiency was approximately 80% (Supplementary Fig. 1C). A marked decrease in the protein levels of Mff and an increased expression of BNIP3 were detected in si-p53 transfected groups compared with those in the scrambled siRNA groups (Fig. 7b). To further verify the changes in mitochondrial fission and BNIP3-mediated mitophagy, mitochondria were stained with mito-tracker red. The average length of mitochondria in p53 knockdown group was greater than that in the transfection control group (Fig. 7c). The co-localization of autophagosomes (LC3-II green) and mitochondria (TOMM20 red) was also increased after p53 inhibition (Fig. 7d). These results confirmed our hypothesis that the NR4A1/DNA-PKcs/p53 pathway regulates both Drp1-mediated mitochondrial fission and BNIP3-related mitophagy.
Discussion
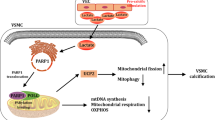
In this study, we explored the characteristics and potential novel mechanisms of mitochondrial homeostasis in vascular calcification, which is correlated with lactate. We found that: (1) lactate accelerated vascular calcification ex vivo, accompanied by excessive mitochondrial fission, suppressed mitophagy, and enhanced apoptosis, (2) NR4A1 was involved in lactate-induced VSMC calcification and contributed to DNA-PKcs and p53 activation, (3) lactate evoked Drp1-related mitochondrial fission but blunted BNIP3-mediated mitophagy, partly through NR4A1, eventually leading to mitochondrial membrane potential decline, VSMC apoptosis, and respiratory dysfunction, and (4) p53 phosphorylation regulated Drp1 and BNIP3, and the NR4A1/DNA-PKcs/p53 axis was involved in impaired mitochondrial homeostasis connected to lactate and vascular calcification. This study further clarifies the specific mechanism by which lactate accelerates vascular calcification, providing a link between the NR4A1/DNA-PKcs/p53 signaling pathway, mitochondrial fission, and mitophagy (Fig. 8).

Summary of the hypothesis of the study. Schematic of this study that lactate exacerbates Drp1-mediated mitochondrial fission via the NR4A1/DNA-PKcs/p53 pathway, causing mitochondrial dysfunction. In addition, lactate inhibits BNIP3-mediated protective mitophagy, eventually leading to increased apoptosis and accelerated vascular calcification
With the increase in the number of patients with diabetes and chronic renal function, vascular tissue calcification has become hindrance to the treatment of cardiovascular diseases, especially for vascular intervention (3). Vascular calcification is advanced atherosclerosis, both of which depend on similar pathological conditions and processes, such as inflammatory stimuli [44], insulin resistance [45], oxidative stress [46], apoptosis [47], and VSMC phenotype transformation [48]. This also explains why vascular calcification and atherosclerosis are significantly associated with inflammatory factors and oxidative stress products. As a by-product of glucose metabolism, lactate has become a recent focus of cardiovascular research. Elevated blood lactate levels suggest a decrease in the flux of tricarboxylic acid cycle, an increase in glucose anaerobic glycolysis, and this metabolic shift is one of the mechanisms of atherosclerotic disease [31]. Our early results show that oxidative stress and apoptosis induced by lactate are important mechanisms of VSMC bone transformation and calcium deposition [38], and the specific mechanism research is the focus of this study.
In rats, vascular calcification can be alleviated by attenuating mitochondrial oxidative stress [49] and endoplasmic reticulum stress-mediated apoptosis [50]. Studies have confirmed that the activation of oxidative stress and apoptosis in vivo is related to the imbalance of mitochondrial fission and mitophagy [16, 51, 52]. Similar to the results at the VSMC level, we confirmed the pro-calcification role of lactate ex vivo. At the same time, we found that mitochondrial over-fission, decreased mitophagy, and increased apoptosis were present in the thoracic aortas following VDN + lactate treatment, which was consistent to the previous studies. Although we did not directly regulate the blood lactate levels in rats, the results of ex vivo experiments could also indicate to some extent that high blood lactate levels will accelerate vascular calcification in vivo. The endothelium and VSMCs of the tunica media are the basis for atherosclerosis and calcification. Especially under pathological conditions, the VSMCs of the medial membrane migrate into the intimal hyperplasia and produce connective tissue, which is an important process in the pathology of arteriosclerosis [53]. Vascular endothelium proliferation, migration, apoptosis, and tube formation are also regulated by mitochondrial fission and mitophagy [54, 55]. Therefore, we speculated that lactate acts on the endothelium as well.
Moderate mitochondrial fission helps the communication of each separate mitochondria; however, pathologic fission impairs the mitochondrial genome, causing the injury of mitochondrial DNA that participates in oxidative phosphorylation and mitochondrial structure [56]. Mitophagy is partially dependent on Drp1-mediated mitochondrial fission [57, 58]. It is generally believed that three signals for mitophagy are initiated: (1) reduced ATP production activates AMP-activated protein kinase (AMPK) to negatively regulate mTOR complex 1 (mTORC1) [59], (2) the ROS/BNIP3 pathway [60], and (3) mPTP opening and mitochondrial swelling [61]. Theoretically, the disruption of mitochondrial structure and oxidative phosphorylation caused by excessive mitochondrial fission can activate these three signaling pathways. In this study, mitochondrial fission was increased in calcified VSMC compared with that in normal VSMC, whereas compensatory increases in mitophagy may be dependent on Drp1-mediated fission. Lactate further promoted mitochondrial fission on the basis of the calcification environment, which caused the mPTP to open, elongated mitochondria to expand, the membrane potential to decrease, insufficient ATP production, dysfunctional respiration, and mitochondrial apoptosis; however, the mitophagy induced by BNIP3 was inhibited, blocking the cellular self-repair mechanism, eventually accelerating VSMC osteoblastic phenotype transition. Autophagy is a continuous process, and it is not yet possible to accurately determine the target of interfering factors. In this study, lactate reduced autophagosome production, and more importantly, blocked autolysosome formation through NR4A1. It has been reported that lactate degrades lysosomes via the ATP6V02/ hypoxia-inducible factor-2α (HIF-2α) pathway [62], however, lysosomal degradation was not observed at the VSMC level by lyso-tracker green staining (data not shown), further indicating that NR4A1 was involved in autophagic flux block.
The DNA-PKcs/p53 signaling pathway is a cellular self-repairing signal to cope with chronic and irreversible damage [63]. However, this repair signal can be disturbed by NR4A1 [28], a transcription factor that regulates the expression of multiple genes. NR4A1 has been reported to be involved in atherosclerosis, especially macrophage inflammatory responses [64], and endothelial dysfunction [26]. Zhou, H. et al. demonstrate that mitochondrial fission and BNIP3-mediated mitophagy are both regulated by NR4A1/DNA-PKcs/p53 axis during nonalcoholic fatty liver disease (NAFLD) [22]. We demonstrated that NR4A1 plays a damaging role in vascular calcification, and as we expected, lactate initiated NR4A1-related signaling pathway, which promoted Drp1-related mitochondrial fission and BNIP3-mediated mitophagy deficiency. Vascular calcification and NAFLD are both oxidative stress and apoptosis implicated, thus, we hypothesized that this signaling pathway initiates oxidative stress and apoptosis under metabolic stress. Moreover, NR4A1 activates gluconeogenesis and increases blood glucose levels. Glucose metabolism is the main breakthrough in VSMC energy acquisition. Approximately 30% of the ATP supply in VSMCs is derived from aerobic glycolysis, and almost 90% of the glycolysis flux contributes to lactate production [65], so there may be a positive cycle of NR4A1 and lactate production.
Our study also has some limitations. The direct effects of lactate on vascular calcification are missing in vivo, and the absence of validation of the experimental signaling pathway in vivo is incomplete in the explanation of the role of NR4A1-mediated signaling pathways in vascular calcification. In addition, verification of endothelial cell levels may be better coordinated with VSMCs to elucidate the specific mechanisms of vascular calcification. Future research will clarify these two issues.
Conclusions
Based on previous experiments, this study not only verified the acceleration of vascular calcification in the presence of lactate ex vivo, but further clarified the role and mechanism of mitochondrial homeostasis regulation in vascular calcification. Lactate exacerbated Drp1-mediated mitochondrial fission via the NR4A1/DNA-PKcs/p53 pathway, causing mitochondria swelling, decreased membrane potential, and disruption of respiratory function. In addition, lactate inhibited BNIP3-mediated mitophagy and compensatory self-clearing, eventually leading to increased apoptosis and accelerated vascular calcification. This study provides evidence of a new mechanism in vascular calcification and highlights the important roles of lactate and mitochondrial homeostasis in clinical vascular diseases.
Abbreviations
- VSMC:
-
Vascular smooth muscle cell
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- RUNX2:
-
Runt-related transcription factor 2
- BMP-2:
-
Bone morphogenetic protein 2
- NR4A1:
-
Nuclear receptor subfamily 4 group A member 1
- DNA-PKcs:
-
DNA-dependent protein kinase, catalytic subunit
- Drp1:
-
Dynamin-related protein 1
- LC3-II:
-
Microtubule-associated protein 1 light chain 3B
- p62:
-
SQSTM1
- TOMM20:
-
Translocase of outer mitochondrial membrane 20
- mPTP:
-
Mitochondrial permeability transition pore
- TEM:
-
Transmission electron microscope
- ATP:
-
Adenosine triphosphate
- OCR:
-
Oxygen consumption rate
References
Dunlay SM, Givertz MM, Aguilar D et al (2019) Type 2 diabetes mellitus and heart failure, a scientific statement from the American Heart Association and Heart Failure Society of America. J Cardiac Fail 50(19):1914–1931
Weng L, Zhang F, Wang R, Ma W, Song Y (2019) A review on protective role of genistein against oxidative stress in diabetes and related complications. Chemico-Biol Interact. https://doi.org/10.1016/j.cbi.2019.05.031
Tolle M, Reshetnik A, Schuchardt M, Hohne M, van der Giet M (2015) Arteriosclerosis and vascular calcification: causes, clinical assessment and therapy. Eur J Clin Invest 45:976–985
Viegas C, Araujo N, Marreiros C, Simes D (2019) The interplay between mineral metabolism, vascular calcification and inflammation in chronic kidney disease (CKD): challenging old concepts with new facts. Aging 11:4274–4299
Zhu Y, Ma WQ, Han XQ, Wang Y, Wang X, Liu NF (2018) Advanced glycation end products accelerate calcification in VSMCs through HIF-1alpha/PDK4 activation and suppress glucose metabolism. Sci Rep 8:13730
Koike S, Yano S, Tanaka S, Sheikh AM, Nagai A, Sugimoto T (2016) Advanced glycation end-products induce apoptosis of vascular smooth muscle cells: a mechanism for vascular calcification. Int J Mol Sci 17:1567
Wang Y, Ma WQ, Zhu Y, Han XQ, Liu N (2018) Exosomes derived from mesenchymal stromal cells pretreated with advanced glycation end product-bovine serum albumin inhibit calcification of vascular smooth muscle cells. Front Endocrinol 9:524
Lee KM, Lee EO, Lee YR et al (2017) APE1/ref-1 inhibits phosphate-induced calcification and osteoblastic phenotype changes in vascular smooth muscle cells. Int J Mol Sci 18(10):2053
Li X, Fang F, Gao Y et al (2019) ROS induced by killerred targeting mitochondria (mtkr) enhances apoptosis caused by radiation via cyt c/caspase-3 pathway. Oxid Med Cell Longev 2019:4528616
Zhang G, Yang W, Jiang F et al (2019) PERK regulates Nrf2/ARE antioxidant pathway against dibutyl phthalate-induced mitochondrial damage and apoptosis dependent of reactive oxygen species in mouse spermatocyte-derived cells. Toxicol Lett 308:24–33
Rovira-Llopis S, Banuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM (2017) Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol 11:637–645
Huang Q, Zhan L, Cao H et al (2016) Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 12:999–1014
Xian H, Liou YC (2019) Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy 20:1–19
Zhou H, Hu S, Jin Q et al (2017) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc 6:e005328
Zhou H, Shi C, Hu S, Zhu H, Ren J, Chen Y (2018) BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis 21:599–615
Rogers MA, Maldonado N, Hutcheson JD et al (2017) Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res 121:220–233
Chan CM, Huang DY, Sekar P, Hsu SH, Lin WW (2019) Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J Biomed Sci 26:40
Ma WQ, Sun XJ, Wang Y, Zhu Y, Han XQ, Liu NF (2019) Restoring mitochondrial biogenesis with metformin attenuates beta-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol Cell Endocrinol 479:39–53
Qi J, Wang F, Yang P et al (2018) Mitochondrial fission is required for angiotensin ii-induced cardiomyocyte apoptosis mediated by a Sirt1-p53 signaling pathway. Front Pharmacol 9:176
Li H, Feng J, Zhang Y et al (2019) Mst1 deletion attenuates renal ischaemia-reperfusion injury: the role of microtubule cytoskeleton dynamics, mitochondrial fission and the GSK3beta-p53 signalling pathway. Redox Biol 20:261–274
Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA (2013) p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 62:70–77
Zhou H, Du W, Li Y et al (2018) Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res 64:e12450
Xing M, Oksenych V (2019) Genetic interaction between DNA repair factors PAXX, XLF, XRCC4 and DNA-PKcs in human cells. FEBS Open Bio 8:426–434
Yan Q, Zhu H, Lan L, Yi J, Yang J (2017) Cleavage of Ku80 by caspase-2 promotes non-homologous end joining-mediated DNA repair. DNA Repair 60:18–28
Koike M, Yutoku Y, Koike A (2011) Accumulation of p21 proteins at DNA damage sites independent of p53 and core NHEJ factors following irradiation. Biochem Biophys Res Commun 412:39–43
Li P, Bai Y, Zhao X et al (2018) NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways. Cell Stress Chaperones 23:749–761
Hanna RN, Shaked I, Hubbeling HG et al (2012) NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res 110:416–427
Zhao BX, Chen HZ, Du XD et al (2011) Orphan receptor TR3 enhances p53 transactivation and represses DNA double-strand break repair in hepatoma cells under ionizing radiation. Mol Endocrinol 25:1337–1350
Yu Y, Cai Z, Cui M et al (2015) The orphan nuclear receptor Nur77 inhibits low shear stress-induced carotid artery remodeling in mice. Int J Mol Med 36:1547–1555
Zhou H, Wang J, Zhu P et al (2018) NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol 113:23
Vallee A, Vallee JN, Lecarpentier Y (2019) Metabolic reprogramming in atherosclerosis: Opposed interplay between the canonical WNT/beta-catenin pathway and PPARgamma. J Mol Cell Cardiol 133:36–46
Yang Q, Xu J, Ma Q et al (2018) PRKAA1/AMPKalpha1-driven glycolysis in endothelial cells exposed to disturbed flow protects against atherosclerosis. Nat Commun 9:4667
Crawford SO, Hoogeveen RC, Brancati FL et al (2010) Association of blood lactate with type 2 diabetes: the atherosclerosis risk in communities carotid MRI study. Int J Epidemiol 39:1647–1655
Shantha GP, Wasserman B, Astor BC et al (2013) Association of blood lactate with carotid atherosclerosis: the atherosclerosis risk in communities (ARIC) carotid MRI study. Atherosclerosis 228:249–255
Juraschek SP, Bower JK, Selvin E et al (2015) Plasma lactate and incident hypertension in the atherosclerosis risk in communities study. Am J Hypertens 28:216–224
Petsophonsakul P, Furmanik M, Forsythe R et al (2019) Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol 39:1351–1368
Yang L, Gao L, Nickel T et al (2017) Lactate promotes synthetic phenotype in vascular smooth muscle cells. Circ Res 121:1251–1262
Zhu Y, Ji JJ, Yang R et al (2019) Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal 58:53–64
Akiyoshi T, Ota H, Iijima K et al (2016) A novel organ culture model of aorta for vascular calcification. Atherosclerosis 244:51–58
Makela J, Tselykh TV, Kukkonen JP, Eriksson O, Korhonen LT, Lindholm D (2016) Peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist is neuroprotective and stimulates PGC-1alpha expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 102:266–275
Andrade MC, Carmo LS, Farias-Silva E, Liberman M (2017) Msx2 is required for vascular smooth muscle cells osteoblastic differentiation but not calcification in insulin-resistant ob/ob mice. Atherosclerosis 265:14–21
Lewis TL Jr, Kwon SK, Lee A, Shaw R, Polleux F (2018) MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat Commun 9:5008
Macchioni L, Petricciuolo M, Davidescu M et al (2018) Palmitate lipotoxicity in enteric glial cells: Lipid remodeling and mitochondrial ROS are responsible for cyt c release outside mitochondria. Biochim Biophys Acta 1863:895–908
Bessueille L, Magne D (2015) Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell Mol Life Sci CMLS 72:2475–2489
Ong KL, McClelland RL, Rye KA et al (2014) The relationship between insulin resistance and vascular calcification in coronary arteries, and the thoracic and abdominal aorta: the multi-ethnic study of atherosclerosis. Atherosclerosis 236:257–262
Beloqui O, Moreno MU, San Jose G et al (2017) Increased phagocytic NADPH oxidase activity associates with coronary artery calcification in asymptomatic men. Free Radical Res 51:389–396
Xia W, Li Y, Wu M et al (2019) Inhibition of mitochondrial activity ameliorates atherosclerosis in ApoE(-/-) mice via suppressing vascular smooth cell activation and macrophage foam cell formation. J Cell Biochem 120:17767–17778
Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM (2018) Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res 114:590–600
Feng H, Wang JY, Yu B et al (2019) Peroxisome proliferator-activated receptor-gamma coactivator-1alpha inhibits vascular calcification through sirtuin 3-mediated reduction of mitochondrial oxidative stress. Antioxid Redox Signal 31:75–91
Shi Y, Wang S, Peng H et al (2019) Fibroblast growth factor 21 attenuates vascular calcification by alleviating endoplasmic reticulum stress mediated apoptosis in rats. Int J Biol Sci 15:138–147
Li MY, Ding JQ, Tang Q et al (2019) SIRT1 activation by SRT1720 attenuates bone cancer pain via preventing Drp1-mediated mitochondrial fission. Biochim Biophys Acta 1865:587–598
Zhang T, Wu P, Budbazar E et al (2019) Mitophagy reduces oxidative stress via Keap1 (Kelch-like epichlorohydrin-associated protein 1)/Nrf2 (nuclear factor-E2-related Factor 2)/PHB2 (prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke 50:978–988
Santulli G (2015) microRNAs distinctively regulate vascular smooth muscle and endothelial cells: functional implications in angiogenesis, atherosclerosis, and in-stent restenosis. Adv Exp Med Biol 887:53–77
Kim DY, Jung SY, Kim YJ et al (2018) Hypoxia-dependent mitochondrial fission regulates endothelial progenitor cell migration, invasion, and tube formation. Korean J Physiol Pharmacol 22:203–213
Jin H, Zhu Y, Li Y et al (2019) BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 24:511–528
Meyer JN, Leuthner TC, Luz AL (2017) Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391:42–53
Li N, Wang H, Jiang C, Zhang M (2018) Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp Cell Res 369:27–33
Cho HM, Ryu JR, Jo Y et al (2019) Drp1-Zip1 interaction regulates mitochondrial quality surveillance system. Mol Cell 73:364–376.e368
Yau WW, Singh BK, Lesmana R et al (2019) Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 15:131–150
O'Sullivan TE, Johnson LR, Kang HH, Sun JC (2015) BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity 43:331–342
Han X, Zhu J, Zhang X et al (2018) Plin4-dependent lipid droplets hamper neuronal mitophagy in the MPTP/p-induced mouse model of Parkinson's disease. Front Neurosci 12:397
Liu N, Luo J, Kuang D et al (2019) Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2alpha-mediated tumor progression. J Clin Investig 129:631–646
Woods DS, Sears CR, Turchi JJ (2015) Recognition of DNA termini by the C-terminal region of the Ku80 and the DNA-dependent protein kinase catalytic subunit. PLoS ONE 10:e0127321
Koenis DS, Medzikovic L, van Loenen PB et al (2018) Nuclear receptor Nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Rep 24:2127–2140.e2127
Kim JH, Bae KH, Byun JK et al (2017) Lactate dehydrogenase-A is indispensable for vascular smooth muscle cell proliferation and migration. Biochem Biophys Res Commun 492:41–47
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 81770451), the Fundamental Research Funds for the Central Universities and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX19_0117), and the Jiangsu Provincial Health and Wellness Committee Research Project (No. H2018001).
Author information
Authors and Affiliations
Contributions
YZ was involved in the experimental design, biochemistry detection, data analysis, and writing of the manuscript. WQM, XQH, SXJ, and YR contributed to the animal model construction and cell culture. NFL was the supervisor and provided expertise in experimental design.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhu, Y., Han, XQ., Sun, XJ. et al. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis 25, 321–340 (2020). https://doi.org/10.1007/s10495-020-01592-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-020-01592-7