
Abstract
Ischemic cardiovascular disease remains one of the leading causes of morbidity and mortality in the world. Proangiogenic therapy appears to be a promising and feasible strategy for the patients with ischemic cardiovascular disease, but the results of preclinical and clinical trials are limited due to the complicated mechanisms of angiogenesis. Facilitating the formation of functional vessels is important in rescuing the ischemic cardiomyocytes. EphrinB2/EphB4, a novel pathway in angiogenesis, plays a critical role in both microvascular growth and neovascular maturation. Hence, investigating the mechanisms of EphrinB2/EphB4 pathway in angiogenesis may contribute to the development of novel therapeutics for ischemic cardiovascular disease. Previous reviews mainly focused on the role of EphrinB2/EphB4 pathway in embryo vascular development, but their role in postnatal angiogenesis in ischemic heart disease has not been fully illustrated. Here, we summarized the current knowledge of EphrinB2/EphB4 in angiogenesis and their interaction with other angiogenic pathways in ischemic cardiovascular disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic cardiovascular disease such as myocardial infarction (MI) is a major cause of morbidity and mortality world widely. Early reperfusion of the occluded coronary arteries potentially improves cardiac function and outcomes by restoring blood supply to the ischemic areas [1, 2]. However, microvascular rarefaction and/or dysfunction prevents efficient reperfusion to the entire myocardium. In this regard, de novo formation of microvessels, namely angiogenesis and arteriogenesis, has the potential to salvage ischemic myocardium at early stages after MI and is also essential for long-term left ventricular remodeling to prevent heart failure [1, 3, 4].
Angiogenesis, the formation of new capillaries from pre-existing blood vessels (Fig. 1a), is essential for transporting oxygen and nutrients to ischemic region and disposing of waste, which has been most extensively studied. Other blood vessel formations, such as vasculogenesis and arteriogenesis (Fig. 1b, c), are also indispensable in physiologic and pathologic neovascularization [5]. In fact, there are at least two different mechanisms of angiogenesis: true sprouting of capillaries from pre-existing vessels termed sprouting angiogenesis and nonsprouting angiogenesis including bridging and intussusceptions [6]. After birth sprouting angiogenesis participates most extensively in vessel formation. In this review, we will focus on the sprouting angiogenesis.

Schematic overview of the three main ways of neovessel formation. a Angiogenesis, new capillaries formation from pre-existing blood vessels by sprouting. b Vasculogenesis, new blood vessel formation by endothelial progenitors. c Arteriogenesis, the formation of the conduit vessels from small collateral arteries. Ultimately, functional mature vessel networks form to support the ischemic region
The Ephrin/Eph system, the largest family of tyrosine kinase receptors in mammals, involves in widespread physiologic and pathologic angiogenesis [7]. Among them, EphrinB2 and its receptor EphB4 play a crucial role in the development of the cardiovascular system and contribute to the function of vasculature [8]. Interference with EphrinB2/EphB4 interactions destabilizes the development of capillary network, and deficiency in EphrinB2 or EphB4 displays similar early embryonic lethality due to disorganized vasculature in mice [9–11]. In this review, we will review the role of EphrinB2/EphB4 in postnatal angiogenesis and their potential role in ischemic cardiovascular disease.
Ephrin/Eph family and structural features
The name “Eph” is an acronym from erythropoietin-producing hepatoma where it was found to be highly expressed at first [12], and its ligand Ephrin is short for erythropoietin-producing hepatoma interactor. There are 14 receptors and 8 ligands in mammals. According to their sequence similarity and binding specificities, both the receptors and ligands can be classified into two categories, A and B [13, 14]. In general, A-type receptors only bind A-type ligands, so do B-type receptors and ligands. But there are exceptions, for instance, EphA4 can bind both A-type and most B-type Ephrins. EphB2, besides the EphrinBs, also binds EphrinA5. However, EphB4 has been currently identified as the specific receptor of EphrinB2 [7, 15, 16].
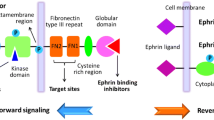
Interaction of the Eph with Ephrin requires cell–cell contact because both the receptor and ligand are membrane-bound, resulting in bidirectional signaling. Eph-activated signaling is termed “forward,” and signaling induced by the Ephrin is named “reverse” [14, 17, 18]. Both EphA and B receptors have similar structures consisting of the extracellular portion, a single transmembrane region and the intracytoplasmic portion [19] (Fig. 2a). The extracellular portion consists of a globular ligand-binding domain; an EGF-like, cysteine-rich region; and two fibronectin-type III repeats. The intracellular portion is composed of a juxtamembrane portion, a tyrosine kinase region, a sterile alpha motif (SAM) domain and a PDZ-binding motif. In contrast to Eph, EphrinA and B have different structures. EphrinA that has an Eph receptor-binding domain in the extracellular portion is tethered to the cell membrane via a glycosylphosphatidylinositol (GPI) anchor, and they have no intracytoplasmic domain to allow signal transmission. EphrinB contains an Eph receptor-binding domain, a transmembrane region, a short cytoplasmic portion with several tyrosine and serine phosphorylation sites and a PDZ-binding motif in the C-terminal [20, 21] (Fig. 2b). While contacting Ephrin on adjacent cells, Eph initiates “forward” signaling by auto-phosphorylation of several tyrosine residues of intracytoplasmic tyrosine kinase domain. At the same time, the reverse signaling is mediated by the C-terminal region of EphrinB, either through tyrosine phosphorylation by recruitment of other molecules, such as Src family kinases, or a PDZ-dependent way [22].

The structure of Ephrin/Eph family. a The Eph receptors include an extracellular portion that consists of a globular, a cysteine-rich EGF-like region, and two fibronectin-type III repeats, and an intracellular portion composed of a juxtamembrane region, a tyrosine kinase domain, a SAM domain and a PDZ-binding motif. b EphrinA ligands only have an Eph receptor-binding domain connected to the transmembrane segment via a GPI anchor. EphrinB ligands contain an extracellular Eph receptor-binding domain, a transmembrane region, a short cytoplasmic portion with several tyrosine and serine phosphorylation sites and a PDZ-binding motif in C-terminal. Once engagement with each other, bidirectional signaling is activated with Eph “forward” signaling and EphrinB “reverse” signaling
EphrinB2/EphB4 and angiogenesis
Angiogenesis involves matrix breakdown, endothelial cell (EC) sprouting, branching, pruning, differentiating and recruitment of mural cells, which mainly refers to pericytes and vascular smooth muscle cells (vSMCs), to stabilize the neovasculature, and ultimately establishing a mature circulation system [23, 24]. Postnatal angiogenesis, different from embryonic angiogenesis, participates in numerous pathophysiologic processes, such as ischemic cardiovascular disease, tumorigenesis, wound repair and female reproductive cycle [23, 25].
In the vascular system, both EphrinB2 and EphB4 are expressed on endothelia and mural cells, although each has their own preference, EphrinB2 is mainly on arterial endothelia and mural cells, while EphB4 prefers to venous ECs [26–28]. Compared with classical proangiogenic factors such as VEGF/VEGFR and Ang1/Tie2, EphrinB2/EphB4 appears to have potential advantages: not only promotes sprouting angiogenesis, but also participates in vessel maturation: remodeling and stabilization [29–32].
Sprouting, a coordinating process of endothelial migration and proliferation, involves a large number of molecules, as well as EphrinB2 and EphB4. Stimulation of cultured ECs with soluble dimeric forms of EphrinB and EphB induces forward and reverse signaling to promote sprouting angiogenesis [33]. On one hand, activation of EphB4 forward signaling induces EC migration and proliferation [34, 35]. One study showed EphB4 forward signaling alone could induce sprouting behavior of ECs in vitro [36]. Inhibition of EphB4 forward signaling was sufficient to inhibit VEGF-induced angiogenesis in vivo [37, 38]. On the other hand, stimulation of EC EphrinB2 promotes adhesion, migration, chemotaxis, capillary network formation and sprouting angiogenesis [34, 39, 40]. An important role of EphrinB2 reverse signaling has been shown in sprouting angiogenesis, especially in the regulation of tip cell function [41]. Sawamiphak et al. [42] showed that EphrinB2 clusters localized to tip cell filopodia and their expression was up-regulated by activated VEGFR2, which may be related to the VEGF/VEGFR-Dll4/Notch-EphrinB2 cascade [43, 44]. In a retinal angiogenesis model, EphrinB2 PDZ-signaling-deficient mice (EphrinB2∆V) exhibited a reduced number of tip cells with fewer filopodia extensions, which indicated that PDZ-dependent reverse signaling of EphrinB2 regulated vessel sprouting by promoting tip cell filopodia extension [45]. Their later work revealed that EphrinB2 at the tip cell filopodia promoted VEGFR2 endocytosis, thereby activating VEGF signaling to direct filopodia extension. The PDZ mutant of EphrinB2 in ECs could not regulate the internalization of VEGFR2, leading to impaired migration or proliferation of ECs due to loss of VEGF responsiveness [42, 45].
EphrinB2 and EphB4 are also important in neovascular remodeling [36]. Prior studies suggested that both EphB and EphrinB may act in a bimodal manner being capable of transmitting both proadhesive and antiadhesive signals [46–49], thus avoiding tanglesome network by restricting intermingling of the vessels [50]. EphB4 can switch the vascularization program from sprouting angiogenesis to circumferential vessel growth, meanwhile reducing the permeability of the vessels [51]. Indeed, forward EphB4 signaling could suppress sprouting angiogenesis by interfering negatively with VEGF and angiopoietin-1 signaling [40, 52, 53]. EphrinB2 reverse signaling was also showed to inhibit endothelial sprouting but promote circumferential growth of vessels. Several studies revealed that reverse EphrinB2 signaling induced low microvascular density but large vessel diameter resulting in tumor progression [51, 54, 55]. Blockade of EphrinB signaling could reduce EC assembly into cordlike structures [31]. In addition, studies also showed that EphrinB2 expression could be up-regulated by shear stress, and this may be related to the differentiation and remodeling of blood vessels induced by shear stress [44, 56, 57]. The bimodal roles of EphB4 and EphrinB2 may be related to the different spatio-temporal conditions of the angiogenesis.
In addition to vessel remodeling, EphrinB2 is also involved in vessel stabilization. EphrinB2 is massively expressed in mural cells that cover arteries and veins during mouse development, playing an important role in pericyte function [58, 59]. In mice with pericyte-specific EphrinB2 deletion, microvasculature is insufficiently covered with pericytes, and capillaries in multiple organs are immature, leading to diffuse tissue edema, hemorrhaging. Furthermore, aberrant collagen was deposited around immature capillaries of skin in these mutant neonates. Interestingly, the EphrinB2-deficient pericytes appeared morphologically normal in many of these mutant mice, but they were only loosely attached, showing a scattered distribution and insufficient contacting with ECs resulting in incomplete vessel coverage. Meanwhile, mutant vSMCs showed attachment defects and discontinuous microvessels covering [58]. A study using EphrinB2∆V pericytes showed that mutant pericytes had a decreased capacity of stabilizing the capillaries and stimulating synthesis of type IV collagen, a major component of vascular basement membrane, indicating that PDZ-dependent signaling may be associated with the permeability [21]. However, another study showed inconsistent findings, which reported that knock-in mice expressing a PDZ-mutant EphrinB2 were born normally without apparent blood vascular defects, but exhibited marked defects in lymphatic vessel development [60]. The role and mechanism of EphrinB2 in vessel remodeling need to be further studied.
Integrity maintenance among adjacent ECs is also in favor of vascular stabilization. It was shown that the EphrinB2/EphB4 signaling is necessary in the endothelial integrity maintenance through EC/EC interaction [21]. Genetic mouse models have implicated that fine connections were largely absent in cultured EphrinB2 knockout ECs [61].
The downstream signaling of EphB4 and EphrinB2 in angiogenesis
The roles of EphrinB2/EphB4 bidirectional signaling pathways in angiogenesis have been summarized, but little is known about their downstream signals in angiogenesis. Here we will briefly discuss the downstream cascades of EphB4 receptor and EphrinB2 ligand in angiogenesis, respectively.
Downstream signaling of EphB4 receptor
EphB4-induced EC proliferation is at least in part mediated by PI3K/Akt [35] (Fig. 3a). Inhibition of EphB4 could decrease Akt phosphorylation, thereby inhibiting cell proliferation [62, 63]. Likewise, the blockers of PI3K, Akt, PKG and MEK could inhibit EphB4-induced EC proliferation. However, other main signaling pathways of receptor tyrosine kinases such as Ras, Src and phospholipase Cγ had no effect on the proliferation response [35]. According to the previous studies [64, 65], Akt can specifically phosphorylate endothelial nitric-oxide synthase, which will increase the nitrite production following the stimulation of EphB4, thereby activating cGMP-PKG signaling pathway. Additionally, the signaling downstream of EphB4-PI3K/Akt-eNOs/NO-cGMP/PKG is raf but not Ras-dependent MAPK pathway [35, 66]. Therefore, EphB4-induced proliferation signal may, in part, be mediated through the PI3K/Akt-eNOs/NO-cGMP/PKG-raf/MEK/MAP kinase cascade [35, 67, 68] (Fig. 3a).

Schematic representation of the downstream signaling of EphB4 receptor and EphrinB2 ligand. a EphB4 forward signaling is mediated mainly by PI3K-Akt pathway, followed by different signaling to induce cell migration and proliferation. b EphrinB2 reverse signaling mainly relies on tyrosine phosphorylation and a PDZ-dependent way. On the one hand, with the help of SFK, the phosphorylated-dependent signaling is transduced by binding to Grb4 and STAT3. On the other hand, the PDZ-dependent signaling can be mediated by recruiting PDZ-containing proteins such as PDZ-RGS3, Dvl2 and PTP-BL. PTP-BL may negatively regulate EphrinB phosphorylation via SFK
Compared with EphB4-induced proliferation, PI3K/Akt-eNOs/NO pathway may also participate in EphB4-mediated EC migration. The signaling downstream of NO in EphB4-mediated migration may be via focal adhesion kinase (FAK) signaling (PI3K/Akt-eNOs/NO-FAK) but not via PKG-MEK pathway [35, 69–73]. Other evidences indicated that PI3K/Akt-NFκB-MMP cascade may also participate in EphB4-mediated EC migration [35, 74, 75] (Fig. 3a). However, in human umbilical vein EC (HUVECs) it has been found that stimulation of EphB4 was unable to stimulate Akt or Erk but acted as a suppressor of Ras/MAPK signaling via the recruitment of p120-RasGAP [52, 71, 76, 77]. The inconsistent results may be context-dependent and related to the critical role of EphrinB2/EphB4 system for proper morphogenesis of capillary endothelium, while neovessels contact with each other.
Downstream signaling of EphrinB2 ligand
Distinct from the forward EphB4 signaling, the EphrinB2-mediated reverse signaling relies on recruiting signaling molecules due to lack of intrinsic catalytic activity. On one hand, the phosphorylation of EphrinB2 is mediated by recruitment of Src family kinases (SFK) [33, 78], followed by binding of the SH2/SH3 domain-containing adaptor proteins such as Grb4 and STAT3 [79–81] (Fig. 3b). Grb4 induces a variety of cytoskeleton regulation signaling such as FAK, G-protein-coupled receptor kinase-interacting protein (GIT) 1, dynamin, Cbl-associated protein (CAP/ponsin), the Abl-interacting protein (Abi-1) and p21-activated kinase (PAK1), thereby mediating migration of ECs [80, 81] (Fig. 3b). STAT3 protein transduces EphrinB signaling from the cell membrane to the nucleus, which was found to be involved in the extracellular matrix-mediated assembly of ECs and pericytes induced by EphrinB reverse signaling [78, 82] (Fig. 3b). On the other hand, the PDZ-containing proteins can be recruited to the PDZ-binding motif of EphrinB2 to mediate the phosphorylation-independent signaling, which is rather important in both angiogenic sprouting and neovascular stabilization as aforementioned [21, 42]. Among PDZ-containing proteins, the regulator of G-protein-signaling (PDZ-RGS3) and disheveled-2 (Dvl2) may be related to PDZ-dependent EphrinB2 reverse signaling [60, 83–85]. But the downstream effector proteins are obscure at present. The PDZ-RGS3 may partly mediate EphrinB2 reverse signaling by regulating the G-protein-signaling pathway [83, 86] (Fig. 3b). The true mechanisms by which EphrinB2 reverse signaling contributes to angiogenesis require further investigation.
There existed evidences of cross-regulation between phosphorylation and PDZ-dependent EphrinB signaling. For example, phosphotyrosine-dependent signaling of EphrinB firstly occurred via binding to EphB receptor, followed by PDZ-dependent signaling. Additionally, the PDZ-containing protein tyrosine phosphatase PTP-BL was recruited to the activated EphrinB and negatively regulated EphrinB phosphorylation via SFK [33] (Fig. 3b).
Conventionally, EphrinB2 reverse signaling requires cell–cell contact. But some studies also showed that EphrinB2 may have some contact-independent functions, suggesting that reverse signaling can also be triggered in a cell-autonomous receptor-independent fashion [21, 58, 87]. This may be related to the existence and the expression levels of cognate receptors on adjacent cells in different spatio-temporal conditions of the angiogenesis [87]. Gene regulation or cross-talks with other angiogenic molecules may be the potential mechanisms involved. Consistent with the notion, several studies showed that EphrinBs can also become phosphorylated without EphB4 engagement, but interacting with some growth factors receptors, such as FGF, PDGF, EGFR, TIE2 receptor [17, 36, 88–90]. However, more studies will be needed to elucidate the precise mechanisms in this field.
VEGF-Dll4/Notch-EphrinB2 cascade
Vascular endothelial growth factor (VEGF) is one of the most powerful angiogenesis activators [91] and promotes EC expression of EphrinB2, but not its phosphorylation [14, 32, 50]. The Dll4/Notch induced by VEGF [92, 93] is another indispensable pathway in neovascularization [94] and can selectively promote the expression of EphrinB2 [43, 44], suggesting that there may exist a cascade among VEGF–Dll4/Notch-EphrinB2 in angiogenesis [43, 44]. VEGF was reported to induce Notch/Delta-directed specific signaling through the PI3K/Akt pathway, which may be related to the Foxc transcription factors [95]. As reported previously, RBPJ protein is the transcriptional mediator of Notch signaling and may be involved in the expression of EphrinB2 [96]. Therefore, VEGF may induce the expression of EphrinB2 in VEGF/VEGFR-PI3K/Akt-Foxc-Dll4/Notch-RBPJ-EphrinB2 cascade (Fig. 4).

Proposed pathways relative to VEGF/VEGFR-Dll4/Notch-EphrinB2 cascade. VEGF can not only stimulate EC proliferation and migration directly in different pathways, but also induce the expression of EphrinB2 via VEGF/VEGFR-Dll4/Notch-EphrinB2 cascade, thereby inducing the bidirectional signaling of EphrinB2/EphB4. Also, several feedback loops have been shown in the sketch. The diagram here presented is as simplification of the complex pathways related to the VEGF/VEGFR-Dll4/Notch-EphrinB2 cascade. Moreover, many links between the arrows need to be confirmed
Dll4 blockade and soluble EphrinB2 treatment induced nonproductive angiogenesis, characterized by an increase in vascular density but decrease in tissue perfusion, and the effect was additive to that of VEGF [32]. This phenomenon indicates the VEGF-Dll4-EphrinB2 cascade may play a key role in the remodeling of neovessels but not the proliferation of EC [43] (Fig. 4). Activation of EphB kinases suppressed VEGF-induced proliferation and migration through direct inhibition of the Ras/MAPK signaling cascade in ECs, whereas VEGF-induced Flk (VEGFR2) phosphorylation did not alter [52]. Therefore, Dll4 blockade may induce nonproductive angiogenesis, at least in part, by decreasing the negative effect of EphB4 on VEGF-induced EC proliferation [97, 98] (Fig. 4). In line with this notion, knockdown of EphrinB2 with siRNA mimicked the effect of Dll4 blockade [32]. Likewise, the soluble EphrinB2 suppressed VEGF-induced proliferation, sprouting and migration of cultured ECs [99]. Herein, the VEGF-Dll4/Notch-EphrinB2 cascade may inhibit VEGF-induced angiogenesis through EphB4 forward signaling. However, there were some paradoxical results that soluble EphrinB2 not only could inhibit VEGF-induced angiogenesis, but also induce nonproductive angiogenesis [32], and inhibition of EphB4 forward signaling could inhibit VEGF-induced angiogenesis in vivo sufficiently [37]. Agonists of either EphrinB2 or EphB4 were reported to significantly increase the VEGF mRNA levels in an Erk-dependent way, while respective siRNAs for EphrinB2 and EphB4 inhibiting this increase [100]. This may be the reason for the inconsistent outcomes. The divergence may attribute to the distinct functions of the EphrinB2/EphB4 pathway in specific temporal–spatial conditions, such as different cell types, various microenvironment (in vivo or in vitro), distinct stages (sprouting or remodeling) and divergent systems from different laboratories, or the existence of cross-talk between them such as feedback loop and other relevant pathways.
Different from VEGF-induced proliferation, migration of EC is mediated through PI3K/Akt-eNOS/NO-FAK pathway [69, 93], which may share a common PI3K/Akt pathway with the VEGF-Dll4/Notch-EphrinB2 cascade (Fig. 4). The Ras/MAPK pathway is also involved in VEGF-induced migration [52]. These two pathways of PI3K/Akt and Ras/MAPK/ERK signaling can inhibit each other [101, 102]. This may account for proper remodeling of neovessels through the arrest of EC proliferation and migration.
In addition, there may exist some feedback loops in the cascade (Fig. 4). First, a positive-feedback loop may exist in Dll4/Notch signaling of EC [103]. A negative-feedback loop between Dll4/Notch signaling and VEGF signaling was also discovered [104, 105], indicating that Dll4 may act as a “brake” on VEGF-mediated angiogenic sprouting. This feedback loop may partly participate in Dll4 blockade-induced nonproductive angiogenesis. As an effector downstream of VEGF signaling, EphrinB2 in turn affects the activity or expression of VEGF. EphrinB2 can not only promote the endocytosis of VEGFR [42, 61, 106], but also increase the level of VEGF [100], suggesting a positive-feedback loop between EphrinB2 and VEGF/VEGFR. Therefore, the up-regulation of VEGFRs induced by inhibition of EphrinB2 signaling may be the result of a compensation for the decrease of VEGF expression or VEGFR endocytosis [107, 108]. Additionally, it was indicated that two evolutionarily conserved binding sites for the RBPJ protein, the transcriptional mediator of Notch signaling, are present in introns 1 and 2 of the Efnb2 (EphrinB2) gene [96], prompting that the expression of EphrinB2 may in turn affect Notch signaling. Therefore, whether there exist some feedback loops between EphrinB2 and Notch signaling needs further study.
Besides the effect on EphrinB2, VEGF-Dll4/Notch-EphrinB2 cascade also influences the expression of EphB4. VEGF inhibits the expression of EphB4 in adult venous EC directly in a MAPK/ERK-dependent manner [107] (Fig. 4). Notch signaling may exert potent inhibitory effects on EphB4 expression by overexpressing HERPs [103] (Fig. 4). Nevertheless, this effect was not exerted in general [103]. More studies may be required to clarify the mechanisms.
Interestingly, recent studies also showed that Dll4/Notch-EphrinB2 pathway is critically involved in VEGFC/VEGFR3 signaling-induced lymphangiogenesis, which is similar to that involved in VEGF/VEGFR (usually refers to VEGFA/VEGFR2) in angiogenesis. Hence, EphrinB2 may participate in both angiogenesis and lymphangiogenesis, although the structure and function between the blood vessels and lymphatic vasculature are different [60, 61, 109].
EphrinB2/EphB4 and ischemic cardiovascular disease
Angiogenesis is essential for revascularization of ischemic myocardium following infarction. Preliminary clinical study suggests that the therapeutic angiogenesis can provide additional blood flow to the incompletely revascularized areas [110, 111]. EphrinB2/EphB4 pathway may play a pivotal role in modulating the angiogenic process in ischemic cardiovascular disease [19].
Ephrin signaling can be of similar importance as VEGF, and both may function in concert with each other in different cardiac pathologies. EphB4 and EphrinB2 were highly and consistently expressed after 24 h in myocardial infarction, compared with those from the control mice. Furthermore, EC proliferation was increased in the peri-infarcted area post-MI with a tendency at a much greater extent after the EphrinB2-Fc treatment. In noninfarcted mice, treatment with EphrinB2-Fc did not affect the mitotic activity in the myocardium, suggesting that EphrinB2/EphB4 pathway regulates the angiogenesis where new blood vessels are needed, such as the ischemic area, but not affect the normal tissues [112], while the detailed mechanism remains to be uncertain. In cultured human aortic ECs, EphrinB2-Fc induced cell proliferation. In a murine aortic ring angiogenesis model, EphrinB2-Fc was as potent as VEGF in inducing sprout formation [112]. Similarly, in a limb ischemia model, much stronger expression of EphrinB2 was detected in ischemic muscles from growing vessels, compared with that in normal muscles from preexisting vessels from the contralateral limb [50]. In addition, EphrinB2 and EphB4 levels were higher in the brains of hypoxic-ischemic rats [113], revealing that hypoxia may induce angiogenesis partly by up-regulating EphB4 and EphrinB2 expression, while MI or other ischemic diseases occur [114].
Therefore, specific compounds such as EphrinB2-Fc and EphB4-Fc that can mimic the EphB4 forward and EphrinB2 reverse signaling might be a promising therapy to facilitate angiogenic sprouting in early stage of ischemic cardiovascular disease. At the later stage, sprouting angiogenesis may be turned off to avoid excessive immature vessels when the vessel density is enough. Then, neovascular remodeling begins. The role of EphrinB2/EphB4 signaling pathway should be switched to the negative mode to promote neovascular maturation. The combination with other maturation factors such as Ang1 and PDGF is also beneficial. It is worth noting that overexpression of EphrinB2 prevents appropriate EC assembly [42, 61, 78]. Therefore, optimal EphrinB2 expression level is necessary and the exact dose and time windows require to be further studied.
In addition to promote angiogenesis via interaction between endothelia–endothelia and endothelia–pericytes, Eph/Ephrin system may also participate in inflammatory angiogenesis by inflammatory cells and endothelia interaction [115], which may be partly beneficial to the healing of ischemic cardiovascular disease (Fig. 5). Inflammatory cells, such as neutrophils [116], monocytes/macrophages [117, 118], T lymphocytes [119], express Eph/Ephrin molecules and interact with ECs of the vessels surrounding the ischemic area via the EphrinB2/EphB4 pathway to promote angiogenesis [120] (Fig. 5). As mentioned above, EphrinB2 may also participate in the ischemic cardiovascular disease by inducing lymphangiogenesis, which may be useful for reducing edema and thereby relieving interstitial fibrosis after MI [121–123]. Taken together, EphrinB2/EphB4 pathway may be a potential therapeutic target for ischemic cardiovascular disease by generating functional neovasculature and lymphangiogenesis (Fig. 5). More direct roles of EphrinB2/EphB4 in ischemic cardiovascular disease need to be confirmed.

The role of EphrinB2/EphB4 in ischemic cardiovascular disease. Once myocardial infarction (MI) occurs, ischemia and hypoxia may induce the expression of EprinB2/EphB4 in different cells, such as endothelial cell, pericytes and inflammatory cells, which will induce both angiogenesis and lymphangiogenesis signaling and finally promote the healing of MI
Conclusions and perspectives
In recent years, great strides have been made in studying the signaling network linking EphrinB2/EphB4 to angiogenesis. In this review, we summarized the evidences to support the critical role of the B family of Ephs and Ephrins, especially EphrinB2/EphB4, in postnatal angiogenesis and their potential involvement in ischemic cardiovascular diseases. We outlined the individual roles of EphB4 forward and EphrinB2 reverse signaling and their downstream and upstream cascades in angiogenesis, respectively. Nevertheless, our knowledge remains limited and lots of intriguing questions need to be settled: the role of other subtypes of Ephrin/Eph pairs in angiogenesis; the correlations with other RTK families and other proangiogenic factor; and whether the EphrinB/EphB signaling can modulate the electrical coupling of cardiomyocytes through effects on gap junctions [124].
Further investigation should be focused on the precise mechanisms of temporal–spatial regulation, the potential therapies targeting the genes or the molecules of the Eph family members and the development of specific Eph/Ephrin interfering compounds, such as antibodies, receptor and soluble extracellular domain/Fc chimera, which may provide a novel and promising way to treat angiogenic disorders. And a potential therapeutic medicine should be harmoniously promoting the expression of EphrinB2 and EphB4, not just one of them.
References
Shah AM, Mann DL (2011) In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378(9792):704–712. doi:10.1016/S0140-6736(11)60894-5
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357(11):1121–1135. doi:10.1056/NEJMra071667
van der Laan AM, Piek JJ, van Royen N (2009) Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol 6(8):515–523. doi:10.1038/nrcardio.2009.103
Lassaletta AD, Chu LM, Sellke FW (2011) Therapeutic neovascularization for coronary disease: current state and future prospects. Basic Res Cardiol 106(6):897–909. doi:10.1007/s00395-011-0200-1
Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9(6):653–660. doi:10.1038/nm0603-653
Risau W (1997) Mechanisms of angiogenesis. Nature 386(6626):671–674. doi:10.1038/386671a0
Salvucci O, Tosato G (2012) Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv Cancer Res 114:21–57. doi:10.1016/B978-0-12-386503-8.00002-8
Pitulescu ME, Adams RH (2010) Eph/ephrin molecules–a hub for signaling and endocytosis. Genes Dev 24(22):2480–2492. doi:10.1101/gad.1973910
Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F (1987) A novel putative tyrosine kinase receptor encoded by the eph gene. Science 238(4834):1717–1720
Gerety SS, Anderson DJ (2002) Cardiovascular ephrinB2 function is essential for embryonic angiogenesis. Development 129(6):1397–1410
Gerety SS, Wang HU, Chen ZF, Anderson DJ (1999) Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell 4(3):403–414
Murai KK, Pasquale EB (2003) ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci 116(Pt 14):2823–2832. doi:10.1242/jcs.00625
Daar IO (2012) Non-SH2/PDZ reverse signaling by ephrins. Semin Cell Dev Biol 23(1):65–74. doi:10.1016/j.semcdb.2011.10.012
Pasquale EB (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133(1):38–52. doi:10.1016/j.cell.2008.03.011
Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, Vearing C, Geleick D, Feldheim DA, Boyd AW, Henkemeyer M, Nikolov DB (2004) Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci 7(5):501–509. doi:10.1038/nn1237
Brambilla R, Bruckner K, Orioli D, Bergemann AD, Flanagan JG, Klein R (1996) Similarities and differences in the way transmembrane-type ligands interact with the Elk subclass of Eph receptors. Mol Cell Neurosci 8(2–3):199–209. doi:10.1006/mcne.1996.0057
Lee HS, Mood K, Battu G, Ji YJ, Singh A, Daar IO (2009) Fibroblast growth factor receptor-induced phosphorylation of ephrinB1 modulates its interaction with Dishevelled. Mol Biol Cell 20(1):124–133. doi:10.1091/mbc.E08-06-0662
Pasquale EB (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 10(3):165–180. doi:10.1038/nrc2806
Brantley-Sieders DM, Chen J (2004) Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis 7(1):17–28. doi:10.1023/B:AGEN.0000037340.33788.87
Chrencik JE, Brooun A, Recht MI, Kraus ML, Koolpe M, Kolatkar AR, Bruce RH, Martiny-Baron G, Widmer H, Pasquale EB, Kuhn P (2006) Structure and thermodynamic characterization of the EphB4/Ephrin-B2 antagonist peptide complex reveals the determinants for receptor specificity. Structure 14(2):321–330. doi:10.1016/j.str.2005.11.011
Kida Y, Ieronimakis N, Schrimpf C, Reyes M, Duffield JS (2013) EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol (JASN) 24(4):559–572. doi:10.1681/ASN.2012080871
Park I, Lee HS (2015) EphB/ephrinB signaling in cell adhesion and migration. Mol Cells 38(1):14–19. doi:10.14348/molcells.2015.2116
Bikfalvi A (2006) Angiogenesis: health and disease. Ann Oncol 17(Suppl 10):x65–x70. doi:10.1093/annonc/mdl239
Troidl K, Schaper W (2012) Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes/metabolism research and reviews 28(Suppl 1):27–29. doi:10.1002/dmrr.2232
Cochain C, Channon KM, Silvestre JS (2013) Angiogenesis in the infarcted myocardium. Antioxid Redox Signal 18(9):1100–1113. doi:10.1089/ars.2012.4849
Gale NW, Baluk P, Pan L, Kwan M, Holash J, DeChiara TM, McDonald DM, Yancopoulos GD (2001) Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev Biol 230(2):151–160. doi:10.1006/dbio.2000.0112
Bai J, Wang YJ, Liu L, Zhao YL (2014) Ephrin B2 and EphB4 selectively mark arterial and venous vessels in cerebral arteriovenous malformation. J Int Med Res 42(2):405–415. doi:10.1177/0300060513478091
Kuijper S, Turner CJ, Adams RH (2007) Regulation of angiogenesis by Eph-ephrin interactions. Trends Cardiovasc Med 17(5):145–151. doi:10.1016/j.tcm.2007.03.003
Abengozar MA, de Frutos S, Ferreiro S, Soriano J, Perez-Martinez M, Olmeda D, Marenchino M, Canamero M, Ortega S, Megias D, Rodriguez A, Martinez-Torrecuadrada JL (2012) Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 119(19):4565–4576. doi:10.1182/blood-2011-09-380006
Poliakov A, Cotrina M, Wilkinson DG (2004) Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev Cell 7(4):465–480. doi:10.1016/j.devcel.2004.09.006
Salvucci O, de la Luz Sierra M, Martina JA, McCormick PJ, Tosato G (2006) EphB2 and EphB4 receptors forward signaling promotes SDF-1-induced endothelial cell chemotaxis and branching remodeling. Blood 108(9):2914–2922. doi:10.1182/blood-2006-05-023341
Yamanda S, Ebihara S, Asada M, Okazaki T, Niu K, Ebihara T, Koyanagi A, Yamaguchi N, Yagita H, Arai H (2009) Role of ephrinB2 in nonproductive angiogenesis induced by Delta-like 4 blockade. Blood 113(15):3631–3639. doi:10.1182/blood-2008-07-170381
Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R (2002) EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell 9(4):725–737
Maekawa H, Oike Y, Kanda S, Ito Y, Yamada Y, Kurihara H, Nagai R, Suda T (2003) Ephrin-B2 induces migration of endothelial cells through the phosphatidylinositol-3 kinase pathway and promotes angiogenesis in adult vasculature. Arterioscler Thromb Vasc Biol 23(11):2008–2014. doi:10.1161/01.ATV.0000096655.56262.56
Steinle JJ, Meininger CJ, Forough R, Wu G, Wu MH, Granger HJ (2002) Eph B4 receptor signaling mediates endothelial cell migration and proliferation via the phosphatidylinositol 3-kinase pathway. J Biol Chem 277(46):43830–43835. doi:10.1074/jbc.M207221200
Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R (1999) Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev 13(3):295–306
Martiny-Baron G, Holzer P, Billy E, Schnell C, Brueggen J, Ferretti M, Schmiedeberg N, Wood JM, Furet P, Imbach P (2010) The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 13(3):259–267. doi:10.1007/s10456-010-9183-z
Xue C, Chen Y, Huang Z, Ge Y, Wang H, Wang J (2014) EphB4 expression in pterygium is associated with microvessel density. Int J Clin Exp Med 7(11):4008–4015
Huynh-Do U, Vindis C, Liu H, Cerretti DP, McGrew JT, Enriquez M, Chen J, Daniel TO (2002) Ephrin-B1 transduces signals to activate integrin-mediated migration, attachment and angiogenesis. J Cell Sci 115(Pt 15):3073–3081
Hamada K, Oike Y, Ito Y, Maekawa H, Miyata K, Shimomura T, Suda T (2003) Distinct roles of ephrin-B2 forward and EphB4 reverse signaling in endothelial cells. Arterioscler Thromb Vasc Biol 23(2):190–197
Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT (2002) Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev 16(20):2684–2698. doi:10.1101/gad.242002
Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A (2010) Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465(7297):487–491. doi:10.1038/nature08995
Hainaud P, Contreres JO, Villemain A, Liu LX, Plouet J, Tobelem G, Dupuy E (2006) The role of the vascular endothelial growth factor-Delta-like 4 ligand/Notch4-ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res 66(17):8501–8510. doi:10.1158/0008-5472.CAN-05-4226
Masumura T, Yamamoto K, Shimizu N, Obi S, Ando J (2009) Shear stress increases expression of the arterial endothelial marker ephrinB2 in murine ES cells via the VEGF-Notch signaling pathways. Arterioscler Thromb Vasc Biol 29(12):2125–2131. doi:10.1161/ATVBAHA.109.193185
Sawamiphak S, Ritter M, Acker-Palmer A (2010) Preparation of retinal explant cultures to study ex vivo tip endothelial cell responses. Nat Protoc 5(10):1659–1665. doi:10.1038/nprot.2010.130
Carter N, Nakamoto T, Hirai H, Hunter T (2002) EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas). Nat Cell Biol 4(8):565–573. doi:10.1038/ncb823
Zou JX, Wang B, Kalo MS, Zisch AH, Pasquale EB, Ruoslahti E (1999) An Eph receptor regulates integrin activity through R-Ras. Proc Natl Acad Sci USA 96(24):13813–13818
Huynh-Do U, Stein E, Lane AA, Liu H, Cerretti DP, Daniel TO (1999) Surface densities of ephrin-B1 determine EphB1-coupled activation of cell attachment through alphavbeta3 and alpha5beta1 integrins. EMBO J 18(8):2165–2173. doi:10.1093/emboj/18.8.2165
Kullander K, Klein R (2002) Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol 3(7):475–486. doi:10.1038/nrm856
Hayashi S, Asahara T, Masuda H, Isner JM, Losordo DW (2005) Functional ephrin-B2 expression for promotive interaction between arterial and venous vessels in postnatal neovascularization. Circulation 111(17):2210–2218. doi:10.1161/01.CIR.0000163566.07427.73
Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, Hammes HP, Grobholz R, Ullrich A, Vajkoczy P (2006) EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J 25(3):628–641. doi:10.1038/sj.emboj.7600949
Kim I, Ryu YS, Kwak HJ, Ahn SY, Oh JL, Yancopoulos GD, Gale NW, Koh GY (2002) EphB ligand, ephrinB2, suppresses the VEGF- and angiopoietin 1-induced Ras/mitogen-activated protein kinase pathway in venous endothelial cells. FASEB J 16(9):1126–1128. doi:10.1096/fj.01-0805fje
Fuller T, Korff T, Kilian A, Dandekar G, Augustin HG (2003) Forward EphB4 signaling in endothelial cells controls cellular repulsion and segregation from ephrinB2 positive cells. J Cell Sci 116(Pt 12):2461–2470. doi:10.1242/jcs.00426
Noren NK, Lu M, Freeman AL, Koolpe M, Pasquale EB (2004) Interplay between EphB4 on tumor cells and vascular ephrin-B2 regulates tumor growth. Proc Natl Acad Sci USA 101(15):5583–5588. doi:10.1073/pnas.0401381101
Noren NK, Foos G, Hauser CA, Pasquale EB (2006) The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat Cell Biol 8(8):815–825. doi:10.1038/ncb1438
Othman-Hassan K, Patel K, Papoutsi M, Rodriguez-Niedenfuhr M, Christ B, Wilting J (2001) Arterial identity of endothelial cells is controlled by local cues. Dev Biol 237(2):398–409. doi:10.1006/dbio.2001.0383
Obi S, Yamamoto K, Shimizu N, Kumagaya S, Masumura T, Sokabe T, Asahara T, Ando J (2009) Fluid shear stress induces arterial differentiation of endothelial progenitor cells. J Appl Physiol 106(1):203–211. doi:10.1152/japplphysiol.00197.2008
Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH (2006) Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124(1):161–173. doi:10.1016/j.cell.2005.10.034
Korff T, Braun J, Pfaff D, Augustin HG, Hecker M (2008) Role of ephrinB2 expression in endothelial cells during arteriogenesis: impact on smooth muscle cell migration and monocyte recruitment. Blood 112(1):73–81. doi:10.1182/blood-2007-12-128835
Makinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R, Wilkinson GA (2005) PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev 19(3):397–410. doi:10.1101/gad.330105
Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH (2010) Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465(7297):483–486. doi:10.1038/nature09002
Ma X, Luo D, Li K, Liu R, Liu Y, Zhu T, Deng D, Zhou J, Meng L, Wang S, Ma D (2012) Suppression of EphB4 improves the inhibitory effect of mTOR shRNA on the biological behaviors of ovarian cancer cells by down-regulating Akt phosphorylation. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban 32(3):358–363. doi:10.1007/s11596-012-0062-2
Steinle JJ, Meininger CJ, Chowdhury U, Wu G, Granger HJ (2003) Role of ephrin B2 in human retinal endothelial cell proliferation and migration. Cell Signal 15(11):1011–1017
Hernandez-Resendiz S, Palma-Flores C, De Los Santos S, Roman-Anguiano NG, Flores M, de la Pena A, Flores PL, Fernandez GJ, Coral-Vazquez RM, Zazueta C (2015) Reduction of no-reflow and reperfusion injury with the synthetic 17beta-aminoestrogen compound Prolame is associated with PI3 K/Akt/eNOS signaling cascade. Basic Res Cardiol 110(2):464. doi:10.1007/s00395-015-0464-y
Zhang Y, Wang SJ, Han ZH, Li YQ, Xue JH, Gao DF, Wu XS, Wang CX (2014) PI3 K/AKT signaling pathway plays a role in enhancement of eNOS activity by recombinant human angiotensin converting enzyme 2 in human umbilical vein endothelial cells. Int J Clin Exp Pathol 7(11):8112–8117
Hood J, Granger HJ (1998) Protein kinase G mediates vascular endothelial growth factor-induced Raf-1 activation and proliferation in human endothelial cells. J Biol Chem 273(36):23504–23508
Kaur S, Kumar TR, Uruno A, Sugawara A, Jayakumar K, Kartha CC (2009) Genetic engineering with endothelial nitric oxide synthase improves functional properties of endothelial progenitor cells from patients with coronary artery disease: an in vitro study. Basic Res Cardiol 104(6):739–749. doi:10.1007/s00395-009-0039-x
Bir SC, Xiong Y, Kevil CG, Luo J (2012) Emerging role of PKA/eNOS pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovasc Res 95(1):7–18. doi:10.1093/cvr/cvs143
Dimmeler S, Dernbach E, Zeiher AM (2000) Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett 477(3):258–262
Goligorsky MS, Abedi H, Noiri E, Takhtajan A, Lense S, Romanov V, Zachary I (1999) Nitric oxide modulation of focal adhesions in endothelial cells. Am J Physiol 276(6 Pt 1):C1271–C1281
Aslam MI, Abraham J, Mansoor A, Druker BJ, Tyner JW, Keller C (2014) PDGFRbeta reverses EphB4 signaling in alveolar rhabdomyosarcoma. Proc Natl Acad Sci USA 111(17):6383–6388. doi:10.1073/pnas.1403608111
Lee TH, Jung H, Park KH, Bang MH, Baek NI, Kim J (2014) Jaceosidin, a natural flavone, promotes angiogenesis via activation of VEGFR2/FAK/PI3K/AKT/NF-kappaB signaling pathways in endothelial cells. Exp Biol Med 239(10):1325–1334. doi:10.1177/1535370214533883
Jouve N, Bachelier R, Despoix N, Blin MG, Matinzadeh MK, Poitevin S, Aurrand-Lions M, Fallague K, Bardin N, Blot-Chabaud M, Vely F, Dignat-George F, Leroyer AS (2014) CD146 mediates VEGF-induced melanoma cell extravasation through FAK activation. Int J Cancer. doi:10.1002/ijc.29370
Park BK, Zeng X, Glazer RI (2001) Akt1 induces extracellular matrix invasion and matrix metalloproteinase-2 activity in mouse mammary epithelial cells. Cancer Res 61(20):7647–7653
Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J (2001) Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J 15(11):1953–1962. doi:10.1096/fj.01-0198com
Xiao Z, Carrasco R, Kinneer K, Sabol D, Jallal B, Coats S, Tice DA (2012) EphB4 promotes or suppresses Ras/MEK/ERK pathway in a context-dependent manner: implications for EphB4 as a cancer target. Cancer Biol Ther 13(8):630–637. doi:10.4161/cbt.20080
Haupaix N, Stolfi A, Sirour C, Picco V, Levine M, Christiaen L, Yasuo H (2013) p120RasGAP mediates ephrin/Eph-dependent attenuation of FGF/ERK signals during cell fate specification in ascidian embryos. Development 140(21):4347–4352. doi:10.1242/dev.098756
Salvucci O, Maric D, Economopoulou M, Sakakibara S, Merlin S, Follenzi A, Tosato G (2009) EphrinB reverse signaling contributes to endothelial and mural cell assembly into vascular structures. Blood 114(8):1707–1716. doi:10.1182/blood-2008-12-192294
Xu NJ, Henkemeyer M (2009) Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat Neurosci 12(3):268–276. doi:10.1038/nn.2254
Segura I, Essmann CL, Weinges S, Acker-Palmer A (2007) Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat Neurosci 10(3):301–310. doi:10.1038/nn1858
Cowan CA, Henkemeyer M (2001) The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature 413(6852):174–179. doi:10.1038/35093123
Bong YS, Lee HS, Carim-Todd L, Mood K, Nishanian TG, Tessarollo L, Daar IO (2007) ephrinB1 signals from the cell surface to the nucleus by recruitment of STAT3. Proc Natl Acad Sci USA 104(44):17305–17310. doi:10.1073/pnas.0702337104
Lu Q, Sun EE, Klein RS, Flanagan JG (2001) Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell 105(1):69–79
Su Z, Xu P, Ni F (2004) Single phosphorylation of Tyr304 in the cytoplasmic tail of ephrin B2 confers high-affinity and bifunctional binding to both the SH2 domain of Grb4 and the PDZ domain of the PDZ-RGS3 protein. Eur J Biochem/FEBS 271(9):1725–1736. doi:10.1111/j.1432-1033.2004.04078.x
Lu Q, Sun EE, Flanagan JG (2004) Analysis of PDZ-RGS3 function in ephrin-B reverse signaling. Methods Enzymol 390:120–128. doi:10.1016/S0076-6879(04)90008-0
Anger T, Klintworth N, Stumpf C, Daniel WG, Mende U, Garlichs CD (2007) RGS protein specificity towards Gq- and Gi/o-mediated ERK 1/2 and Akt activation, in vitro. J Biochem Mol Biol 40(6):899–910
Bochenek ML, Dickinson S, Astin JW, Adams RH, Nobes CD (2010) Ephrin-B2 regulates endothelial cell morphology and motility independently of Eph-receptor binding. J Cell Sci 123(Pt 8):1235–1246. doi:10.1242/jcs.061903
Chong LD, Park EK, Latimer E, Friesel R, Daar IO (2000) Fibroblast growth factor receptor-mediated rescue of x-ephrin B1-induced cell dissociation in Xenopus embryos. Mol Cell Biol 20(2):724–734
Bruckner K, Pasquale EB, Klein R (1997) Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science 275(5306):1640–1643
Thelemann A, Petti F, Griffin G, Iwata K, Hunt T, Settinari T, Fenyo D, Gibson N, Haley JD (2005) Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol Cell Proteomics 4(4):356–376. doi:10.1074/mcp.M400118-MCP200
Morita S, Furube E, Mannari T, Okuda H, Tatsumi K, Wanaka A, Miyata S (2015) Vascular endothelial growth factor-dependent angiogenesis and dynamic vascular plasticity in the sensory circumventricular organs of adult mouse brain. Cell Tissue Res. doi:10.1007/s00441-014-2080-9
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104(9):3219–3224. doi:10.1073/pnas.0611206104
Liu ZJ, Shirakawa T, Li Y, Soma A, Oka M, Dotto GP, Fairman RM, Velazquez OC, Herlyn M (2003) Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23(1):14–25
Watson O, Novodvorsky P, Gray C, Rothman AM, Lawrie A, Crossman DC, Haase A, McMahon K, Gering M, Van Eeden FJ, Chico TJ (2013) Blood flow suppresses vascular Notch signalling via dll4 and is required for angiogenesis in response to hypoxic signalling. Cardiovasc Res 100(2):252–261. doi:10.1093/cvr/cvt170
Hayashi H, Kume T (2008) Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS ONE 3(6):e2401. doi:10.1371/journal.pone.0002401
Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Perez-Pomares JM, de la Pompa JL (2007) Notch signaling is essential for ventricular chamber development. Dev Cell 12(3):415–429. doi:10.1016/j.devcel.2006.12.011
Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley EJ, Duarte A, Gill PS (2007) Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 109(11):4753–4760. doi:10.1182/blood-2006-12-063933
Sturz A, Bader B, Thierauch KH, Glienke J (2004) EphB4 signaling is capable of mediating ephrinB2-induced inhibition of cell migration. Biochem Biophys Res Commun 313(1):80–88
Zamora DO, Davies MH, Planck SR, Rosenbaum JT, Powers MR (2005) Soluble forms of EphrinB2 and EphB4 reduce retinal neovascularization in a model of proliferative retinopathy. Invest Ophthalmol Vis Sci 46(6):2175–2182. doi:10.1167/iovs.04-0983
Das A, Shergill U, Thakur L, Sinha S, Urrutia R, Mukhopadhyay D, Shah VH (2010) Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest Liver Physiol 298(6):G908–G915. doi:10.1152/ajpgi.00510.2009
Blum S, Issbruker K, Willuweit A, Hehlgans S, Lucerna M, Mechtcheriakova D, Walsh K, von der Ahe D, Hofer E, Clauss M (2001) An inhibitory role of the phosphatidylinositol 3-kinase-signaling pathway in vascular endothelial growth factor-induced tissue factor expression. J Biol Chem 276(36):33428–33434. doi:10.1074/jbc.M105474200
Hong CC, Kume T, Peterson RT (2008) Role of crosstalk between phosphatidylinositol 3-kinase and extracellular signal-regulated kinase/mitogen-activated protein kinase pathways in artery-vein specification. Circ Res 103(6):573–579. doi:10.1161/CIRCRESAHA.108.180745
Iso T, Maeno T, Oike Y, Yamazaki M (2006) Doi H, Arai M, Kurabayashi M Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun 341(3):708–714. doi:10.1016/j.bbrc.2006.01.020
Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444(7122):1032–1037. doi:10.1038/nature05355
Li JL, Harris AL (2009) Crosstalk of VEGF and Notch pathways in tumour angiogenesis: therapeutic implications. Front Biosci 14:3094–3110
Gaengel K, Betsholtz C (2013) Endocytosis regulates VEGF signalling during angiogenesis. Nat Cell Biol 15(3):233–235. doi:10.1038/ncb2705
Yang C, Guo Y, Jadlowiec CC, Li X, Lv W, Model LS, Collins MJ, Kondo Y, Muto A, Shu C, Dardik A (2013) Vascular endothelial growth factor-A inhibits EphB4 and stimulates delta-like ligand 4 expression in adult endothelial cells. J Surg Res 183(1):478–486. doi:10.1016/j.jss.2013.01.009
Dimova I, Hlushchuk R, Makanya A, Styp-Rekowska B, Ceausu A, Flueckiger S, Lang S, Semela D, Le Noble F, Chatterjee S, Djonov V (2013) Inhibition of Notch signaling induces extensive intussusceptive neo-angiogenesis by recruitment of mononuclear cells. Angiogenesis 16(4):921–937. doi:10.1007/s10456-013-9366-5
Katsuta H, Fukushima Y, Maruyama K, Hirashima M, Nishida K, Nishikawa S, Uemura A (2013) EphrinB2–EphB4 signals regulate formation and maintenance of funnel-shaped valves in corneal lymphatic capillaries. Invest Ophthalmol Vis Sci 54(6):4102–4108. doi:10.1167/iovs.12-11436
Hanawa K, Ito K, Aizawa K, Shindo T, Nishimiya K, Hasebe Y, Tuburaya R, Hasegawa H, Yasuda S, Kanai H, Shimokawa H (2014) Low-intensity pulsed ultrasound induces angiogenesis and ameliorates left ventricular dysfunction in a porcine model of chronic myocardial ischemia. PLoS ONE 9(8):e104863. doi:10.1371/journal.pone.0104863
Sergienko IV, Masenko VP, Semenova AE, Gabrusenko SA, Naumov VG, Belenkov IuN (2009) Effect of myocardial revascularization on dynamics of factors of angiogenesis in patients with ischemic heart disease. Kardiologiia 49(12):4–10
Mansson-Broberg A, Siddiqui AJ, Genander M, Grinnemo KH, Hao X, Andersson AB, Wardell E, Sylven C, Corbascio M (2008) Modulation of ephrinB2 leads to increased angiogenesis in ischemic myocardium and endothelial cell proliferation. Biochem Biophys Res Commun 373(3):355–359. doi:10.1016/j.bbrc.2008.06.036
Zhu L, Qian L, Wang S, Wang T, Jiang L (2014) Expression of ephrinB2 and EphB4 in a neonatal rat model of periventricular white matter damage. J Perinat Med. doi:10.1515/jpm-2014-0096
Vihanto MM, Plock J, Erni D, Frey BM, Frey FJ, Huynh-Do U (2005) Hypoxia up-regulates expression of Eph receptors and ephrins in mouse skin. FASEB J 19(12):1689–1691. doi:10.1096/fj.04-3647fje
Liu H, Devraj K, Moller K, Liebner S, Hecker M, Korff T (2014) EphrinB-mediated reverse signalling controls junctional integrity and pro-inflammatory differentiation of endothelial cells. Thromb Haemost 112(1):151–163. doi:10.1160/TH13-12-1034
Schruefer R, Sulyok S, Schymeinsky J, Peters T, Scharffetter-Kochanek K, Walzog B (2006) The proangiogenic capacity of polymorphonuclear neutrophils delineated by microarray technique and by measurement of neovascularization in wounded skin of CD18-deficient mice. J Vasc Res 43(1):1–11. doi:10.1159/000088975
Yuan K, Jin YT, Lin MT (2000) Expression of Tie-2, angiopoietin-1, angiopoietin-2, ephrinB2 and EphB4 in pyogenic granuloma of human gingiva implicates their roles in inflammatory angiogenesis. J Periodontal Res 35(3):165–171
Yuan K, Hong TM, Chen JJ, Tsai WH, Lin MT (2004) Syndecan-1 up-regulated by ephrinB2/EphB4 plays dual roles in inflammatory angiogenesis. Blood 104(4):1025–1033. doi:10.1182/blood-2003-09-3334
Yu G, Luo H, Wu Y, Wu J (2003) Ephrin B2 induces T cell costimulation. J Immunol 171(1):106–114
Zamora DO, Babra B, Pan Y, Planck SR, Rosenbaum JT (2006) Human leukocytes express ephrinB2 which activates microvascular endothelial cells. Cell Immunol 242(2):99–109. doi:10.1016/j.cellimm.2006.10.001
Sun QN, Wang YF, Guo ZK (2012) Reconstitution of myocardial lymphatic vessels after acute infarction of rat heart. Lymphology 45(2):80–86
Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, Furth PA, Dworatzek E, Regitz-Zagrosek V (2014) Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther 5(1):153. doi:10.4172/2157-7013.1000153
Laine GA, Allen SJ (1991) Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res 68(6):1713–1721. doi:10.1161/01.RES.68.6.1713
Ishii M, Mueller I, Nakajima T, Pasquale EB, Ogawa K (2011) EphB signaling inhibits gap junctional intercellular communication and synchronized contraction in cultured cardiomyocytes. Basic Res Cardiol 106(6):1057–1068. doi:10.1007/s00395-011-0219-3
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81270179 and 81470384 to M.X.X.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Rights and permissions
About this article

Cite this article
Yang, D., Jin, C., Ma, H. et al. EphrinB2/EphB4 pathway in postnatal angiogenesis: a potential therapeutic target for ischemic cardiovascular disease. Angiogenesis 19, 297–309 (2016). https://doi.org/10.1007/s10456-016-9514-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-016-9514-9