
Abstract
A high proportion of primary percutaneous coronary interventions performed in the setting of acute myocardial infarction, concur with inadequate myocardial perfusion at the microvascular level. This phenomenon, known as “no-reflow” contributes to reperfusion injury, poor prognosis and to unfavorable clinical outcome. In this study, we evaluated the hypothesis that the synthetic 17β-aminoestrogen Prolame, may confer cardioprotection and prevent against no-reflow. In an open-chest model of 30-min ischemia and 90-min reperfusion, male Wistar rats were randomly assigned to different groups: Control, Prolame, Prolame followed by the nitric oxide synthase inhibitor (l-NAME), and 17β-estradiol. Areas of risk, infarct size and no-reflow were determined by planimetry with triphenyltetrazolium chloride and thioflavin-S stains. Structural damage of the vasculature was measured as capillary compression in clarified tissue after intra-atrial injection of Microfil. Hemodynamic function was obtained at the end of stabilization, ischemia and reperfusion; nitric oxide (NO·) content was determined indirectly using the Griess reaction. Activation of the eNOS signaling cascade was determined by western blot. Prolame reduced the infarcted area, decreased the zones of no-reflow and capillary compression by activating the PI3K/Akt/eNOS signaling pathway in correlation with NO· increase. Prolame also activated endothelial cells augmenting NO· production, which was inhibited by ICI182780 (a selective estrogen receptor down-regulator), supporting the notion that the cardioprotective effect of Prolame involves the preservation of endothelium through the activation of estrogen receptor downstream signaling. Our results provide evidence that Prolame has potential therapeutic application in patients with AMI, as it prevents from both vascular and cardiac tissue damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Retrospective and observational studies indicate that primary percutaneous coronary intervention (PCI), the preferred treatment against acute myocardial infarction (AMI), renders satisfactory epicardial coronary flow and greater patency rates of the infarct-related artery [44]. However, PCI does not always guarantee clinical improvement; studies of myocardial contrast echocardiography (MCE) have shown diminished patency rates or even no-perfusion at the coronary microvascular level in many of these patients, condition known as no-reflow (NR) phenomenon which contributes to myocardial reperfusion injury. Trials on primary PCI show that up to 40 % cases concurred with inadequate myocardial perfusion due to microvascular obstruction, which was associated with increased 30-day mortality if not adequately treated (32 vs. 2.8 %) [10, 30].
The factors associated with the establishment of NR include: endothelial dysfunction, compression of capillaries by swollen myocytes, alteration of the vasoregulation pathways, epicardial spasm, mechanical obstruction from embolization, extrinsic coagulation pathways, leukocyte adherence, microvascular ischemia, edema and vasoconstriction mediators [15, 23, 38]. Endothelial cell injury occurs in approximately 20 % of vessels after 60 min of reperfusion and in 40 % of vessels at 20–80 min of reperfusion. Indeed, pioneer reports showed tightly packed erythrocytes and endothelial gaps plugged by platelet and fibrin thrombi with numerous extra vascular red blood cells in capillaries from hearts reperfused only during 20 min [29].
Mechanical thrombectomy applied at the time of catheterization is used as a therapy against no-reflow; however, the administration of agents which increases nitric oxide (NO·) levels in the post-ischemic heart is also a common practice, due to its multiple effects on the cardiovascular system [24]. NO· inhibits platelet aggregation [27], reduces oxygen consumption [33], regulates directly or indirectly myocardial contractility [3], scavenges superoxide anions (O2−) [2], prevents leukocyte adhesion [34] and mediates the anti-proliferative/anti-inflammatory response [11]. In spite of this, the administration of NO· donors like nitroglycerin [4], statins [25], verapamil [52], sodium nitroprusside [1] and abciximab have rendered inconclusive results at the microvascular level. Similar outcomes have been reported for adenosine, which despite of its proved efficacy in numerous experimental models, does not provided maximal coronary vasodilatation [20] neither improved TIMI flow rate in patients with acute ST-segment elevation myocardial infarction [8].
Other approaches have focused on the effects of estrogen at vascular level. In animal models and in patients, estrogen promotes vasodilatation via nitric oxide production, regulates blood pressure, decreases vascular inflammation/atherosclerosis and improves vascular reactivity. The fast vascular response observed after estrogen stimulation suggests the activation of non-genomic mechanisms via membrane receptors and downstream cascade PI3K/Akt/eNOS (phosphoinositide 3-kinase/protein serine-threonine kinase/endothelial nitric oxide synthase) [37].
Here we report on a small molecule that might potentiate the effects of estrogen and NO· production on vasoregulation of post-ischemic hearts. The synthetic 17β-aminoestrogen (AE) Prolame [17β-(3-hydroxy-1-propylamino)-1,3,5(10)-estratrien-3-ol)] is an estradiol analog in which the C17 position of the steroid nucleus is substituted by an amino-alcohol side chain-NH-(CH2)3-OH with three methylenes groups [6] (Supplementary Figure 1). Previous studies have shown that the 17β-AEs, besides its antiplatelet properties have high affinity to estrogen α (ERα) receptor [26], producing changes in vasoregulation [36]. In particular, it was demonstrated that Prolame enhanced NO· production in endothelial cells, platelets and in vivo mouse models [12]. This fact and the recent proposal that antiplatelet agents may be cardioprotective following myocardial infarction by mechanisms not mediated by reduction of microvascular obstruction [43], led us to evaluate whether the 17β-aminoestrogen Prolame might diminish the no-reflow phenomenon and provide cardioprotection in rats with acute myocardial infarction followed by reperfusion.
Methods
Reagents
Chemicals were of reagent or higher grade from Sigma-Aldrich (St Louis, MO) unless otherwise specified. Anti-PI3K monoclonal antibody; polyclonal anti-PHO-PI3K, Tyr458/Tyr199; polyclonal anti-Akt; monoclonal anti-PHO-Akt, Thr308; monoclonal anti-PHO-eNOS, Ser1177; polyclonal anti-NOS and specific PI3K inhibitor LY294002 were all purchased from Cell Signaling Technology Inc. (Danver, MA). The enhanced chemiluminescence detection system was from Millipore Corporation (Bedford, MA) and alkaline phosphatase (AP)-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals used were of the highest purity available from Baker Co. (México) and Sigma-Aldrich (México).
Ethical approval
This investigation was performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the United States National Institutes of Health (US-NIH) (NIH publication 85-23, 1985) and approved by the Ethics Committee of the National Institute of Cardiology I. Ch. Experimental work followed the guidelines of the Norma Oficial Mexicana for the use and care of laboratory animals (NOM-062-ZOO-1999) and for the disposal of biological residues (NOM-087-ECOL-1995).
Experimental preparation
The study was performed with male Wistar rats weighting between 400 and 450 g that were anesthetized with 40 mg/kg sodium pentobarbital. A midline incision was made in the neck, and tracheotomy was performed. The rats were mechanically ventilated with room air supplemented with low-flow oxygen using a small-animal ventilator (Harvard apparatus, Holliston, MA) at a rate of 60 breaths per minute and a tidal volume of 1 ml/100 g body weight and respiratory ratio 1:3. After left thoracotomy, hemodynamic parameters were measured with an SPR-869 Mikro-Tip pressure–volume catheter (Millar Instruments, Houston, TX) introduced into the left ventricle, whereas electrocardiogram registers were obtained using standard limb electrodes. Then, a 6-0 nylon suture was placed around the proximal left coronary artery and the ends were passed through a small plastic tube to form a snare. The artery was occluded by pulling the snare, which was kept in place with a haemostatic clamp. Myocardial ischemia was confirmed by visual cyanosis and maintained during 30 min, then the snare was released and the ischemic myocardium was reperfused for 90 min. Sham rats were subjected to the same surgical protocols performed in ischemic-reperfused rats, except that the snare was not tied.
Heart rate (HR), left ventricular end-diastolic pressure (LVEDP), left ventricular end systolic pressure (LVESP) and maximum change rate of left ventricular pressure rise and fall (±dp/dt) were monitored continuously during the entire ischemic/reperfusion protocol. Data acquisition were recorded with the MPVS Ultra Foundation System (ADInstruments, Spechbach, Germany) and analyzed with the LabChart Pro Software (ADInstruments, Spechbach, Germany).
Experimental groups
Animals were randomly divided into six groups: (1) Sham group, rats without ligation and only threading; (2) Control group, rats with 30 min of ischemia by ligation of the left anterior descending coronary artery and 90 min of reperfusion without any treatment; (3) Prolame group (Pro), rats that received 75 µg/kg Prolame as an intravenous bolus 5 min before reperfusion through the tail veins; (4) Prolame + l-NAME group (Pro + l-NAME), rats to which a single dose of 10 mg/kg of N(G)-nitro-l-arginine methyl ester (l-NAME) was given 5 min after Prolame; (5) 17β-estradiol group, rats that received 12.5 µg/kg of 17β-estradiol as an intravenous bolus 5 min before reperfusion through tail veins and (6) Pro + LY group, in which some rats received the PI3K inhibitor LY294002 (0.5 mg/kg) (Supplementary Figure 2).
Chemical synthesis
17β-(3-Hydroxy-1-propylamino)-1,3,5(10)-Estratrien-3-ol, Prolame, was synthesized from estrone. Chemical purity was established by spectral (IR/NMR/MS) and chromatographic (HPLC, TLC) techniques as previously reported [6].
Measurements of area at risk and infarct size
Measurements of the myocardial area at risk (AAR) and infarct area (IA) were performed according to a previous report [32]. In brief, Evans blue dye was injected into the left atrium to determine ligation area and then, the rats were euthanized by injecting 2 ml of 15 % potassium chloride via femoral vein. After excision, the heart was placed in cold saline and the heart was cut into six slices parallel to the atrio-ventricular groove. The area unstained by Evans blue, indicating AAR was traced in visible light. The extent of myocardial necrosis was evaluated incubating the left ventricular slices in 1 % triphenyltetrazolium chloride (TTC) phosphate buffered saline for 25 min at 37 °C. The outlines of the slices and TTC negative staining (infarct area) were traced and photographed in color.
Area of no-reflow (ANR) and capillary compressions (CC) measurements
At the end of the experimental protocols a group of three rats received a 2 % solution of the perfusion marker thioflavin-S (1 ml/kg) through femoral artery. After 15 min, the animals were euthanized and the hearts excised. Thioflavin-S distribution was visualized and photografied under ultraviolet light (λ = 365 nm).
Some animals from each group (n = 6) were subjected to intra-atrial injection with Microfilm to evaluate the presence of capillary compressions in the left ventricles, as described by Coral-Vázquez et al. [5]. Briefly, a bilateral sternum incision was performed to expose the left ventricle and 3 ml of Microfil, a silicon rubber (Flow Tech., Carver, MA) were perfused into the left ventricle. After contraction stopped, the hearts were rapidly excised and maintained in ice for about 20 min. Afterwards, the hearts were fixed in 10 % formaldehyde for 24 h and cardiac tissue was sectioned into 2-mm-thick transverse cross sections. The tissues were subsequently cleared by sequential 24-h immersions in 25, 50, 75, 95 % and finally 100 % ethanol. Finally, the hearts were placed in pure methyl salicylate for 24–48 h. All steps were carried out at room temperature. CC visualization and quantification were performed under transillumination at 10× magnification in 45 non-adjacent microscopic fields. Total number of CC per left ventricle was measured and compared between the experimental groups.
NO· production in cardiac homogenates and in human umbilical vein endothelial cells (HUVEC)
Left ventricle samples (100 mg) were obtained from each animal. Tissues were added to 300 µl of phosphate buffered saline (PBS: 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, pH 7.4) and homogenized until complete homogenization. The homogenates were then centrifuged at 10,000 rpm for 10 min at 4 °C and the supernatants placed in sterile 1 ml Eppendorf tubes. Later, the resulting supernatants were ultra-filtrated through two filters of 0.45-µm pore size (Ministart, Sartorius Ltd., Gottingen, Germany) and 10 kDa cut-off (Vavispin 2, Sartorius Ltd., Stonehouse, UK), respectively, to eliminate proteins from each sample. Cadmium-coated granules were prepared as described [45] and used within 10 min to reduce nitrates to nitrites. Nitrite (NO2 −) total levels were measured using the colorimetric Greiss reagent according to Granger’s method as indicator of NO· production [13].
Human umbilical cords obtained from the Obstetric/Gynecology Service of the Luis Castelazo Ayala Hospital of the Mexican Institute of Social Security, were immediately placed in 0.9 % NaCl supplemented with an antibiotic and antimycotic cocktail. Primary HUVEC cultures were isolated, grown, and identified in endothelial basal medium EBMTM (Lonza, Walkersville, MD, USA), supplemented with EGMTM SingleQuotTM growth factors in a humidified atmosphere of 5 % CO2 and 95 % O2 at 37 °C. Seven hours before the experiments, HUVEC were washed with phenol red-free Hanks salt solution and kept in phenol red-free DMEM plus 1 % of fetal bovine serum and 1 % of antibiotic–antimycotic cocktail. Later, confluent HUVEC monolayers were incubated for 1 h in phenol red-free Hanks salt solution supplemented with 4 mM l-arginine (Sigma-Aldrich Chemical Co., St. Louis, MO, USA), and then stimulated with 1 µM bradykinin (BK) (Sigma-Aldrich Chemical Co., St. Louis, MO, USA) or with 10 µM Prolame for 15 min. In some experiments HUVEC were incubated with the selective estrogen receptor (ER) down-regulator ICI182780 (Abcam, Cambridge, MA, USA) at 5 µM for 24 h before the addition of Prolame. Culture supernatants were used to measure NO· levels and whole cell extracts obtained with RIPA buffer for protein determination. Confluent cell cultures were typically assayed on passages 5–10.
Western blot analysis
Samples of 100 mg from at least six hearts from each group were individually prepared by adding 1 ml of ice cold RIPA lysis buffer (20 mM Tris HCl, pH 7.5, 150 mM NaCl, 0.1 % SDS, 0.5 % sodium deoxycholate) and 10 µl phenylmethylsulfonyl fluoride (1 mM). Aliquots of samples (60 µg of protein) were separated by 8–10 % SDS-PAGE, electrotransferred onto a cellulose acetate membrane and blocked with 5 % nonfat milk in TBS-T buffer (20 mM Tris HCl, pH 7.4, 135 mM NaCl, 0.1 % Tween-20). Anti-PHO-PI3K, Tyr458/Tyr199 (1:1,000 dilution); anti-PI3K (1:1,000 dilution); polyclonal anti-Akt (1:1,000 dilution); anti-PHO-Akt, Thr 308 (1:1,000 dilution); anti-PHO-eNOS, Ser 1177 (1:1,000 dilution) and anti-NOS (1:1,000 dilution) were used to evaluated the PI3K/Akt/eNOS pathway activation. Secondary antibodies conjugated with alkaline phosphatase were used to detect protein content along with a chemiluminescence detection system (Millipore, Billerica, MA, USA). Autoradiographic images were analyzed using scanning densitometer software. Ratio between phosphorylated protein and total protein was obtained in the same membrane in all experiments, and then data were compared among groups.
Effect of Prolame on impedance aggregometry
Aggregation was determined by measuring impedance with a whole blood aggregometer (Model 560CA, Chrono-log Corporation Havertown, PA, USA). Samples of 0.5 ml heparinized arterial blood (68 USP units) from Sham, Control, Pro and Pro + LY rats were collected. For each assay, whole blood was diluted 1:1 with saline solution (0.9 % w/v) and incubated during 3 min at 37 °C in a plastic cuvette under continuous stirring. Aggregation was stimulated with 10 μM ADP or 2 μg/mL collagen (Chrono-PAR Corporation Havertown, PA; USA) as described by Yang et al. [51]. Platelet aggregation was measured during 6 min and maximum changes were recorded. The data were analyzed with the AggroLink software package.
Statistical analysis
All data are expressed as mean ± SE. Data from all conditions, such as hemodynamic data and other time-dependent determinations, were compared by repeated-measures ANOVA followed by post hoc analysis with Student–Newman–Keuls multiple comparisons. Differences in a single variable, such as ANR and IA, were compared among groups by one-way ANOVA followed by Duncan’s post hoc test. P < 0.05 was considered statistically significant.
Results
Hemodynamic data
HR, LVESP, LVEDP, and ±dp/dt before ischemia (stabilization), at the end of ischemia (Ischemia30 min) and at the end of reperfusion (Reperfusion90 min) were evaluated in all experimental groups (Table 1). No significant differences were observed after the stabilization period in any of these parameters between the groups; neither during 120 min of continuous registers in Sham rats (not shown). After 30 min of ischemia, HR, LVESP and ±dp/dt diminished, whereas LVEDP increased in all groups, as compared with values obtained during stabilization. At the end of reperfusion (Reperfusion90 min), hemodynamic parameters were similar to those observed after the ischemic period (Ischemia30 min) in the Control group. In contrast, HR, LVESP and ±dp/dt values increased and LVEDP diminished in the Pro group. Those changes were significantly different to values obtained in the Control group after reperfusion (P < 0.05). In the Pro + l-NAME group, the cardioprotective effect of Pro was abolished, as well as in the Pro + LY group indicating the participation of the PI3K/Akt/eNOS signaling cascade. On the other hand, 17β-estradiol showed similar effects that those exerted by Prolame.
AAR, IA and ANR evaluation
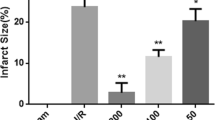
The AAR in the left ventricle (LV) was comparable in Control, Pro, Pro + l-NAME and 17β-estradiol groups, averaging 43 %. The IA observed in the Control group after reperfusion (Reperfusion90 min) was 55 %, whereas in Pro and 17β-estradiol groups diminished to 35 and 45 %, respectively. IA was also significantly lower in the Pro group than in the Pro + l-NAME group (35 ± 3.1 vs. 53 ± 3.6, P < 0.05).
The ANR diminished significantly in the Pro group as compared with the Control, Pro + l-NAME and 17β-estradiol groups (P < 0.05). Coronary vessels were in general smoothly tapered in the Sham group and CC quantification was only of 9.2 ± 2.3/LV. In contrast, CC increased to 79 ± 4.6/LV in the control group in correlation with generalized sparseness of perfusion. Prolame treatment (Pro group) decreased CC to 23 ± 3.8/LV and 17β-estradiol only to 40 ± 2.5/LV. Diminution in CC observed in the Pro group was abolished in presence of l-NAME (63 ± 4.2/LV) (Figs. 1a, b).

Area at risk (AAR), infarct area (IA), area of no-reflow (ANR) and capillary compressions (CC) in reperfused rat hearts treated with Prolame. a Representative images of heart sections stained with: Evans blue, in which unstained zones indicated AAR (panels 1–5); triphenyltetrazolium chloride (TTC) in which pale zones represented IA (panels 6–10) and, thioflavin-S in which the NR area is negative for fluorescence (panels 11–15). Transillumination of Microfil-perfused coronary arteries showing capillary compressions (panels 16–20). The yellow arrow indicates one capillary compression. b Statistical analysis of AAR, IA and ANR expressed in percentage. c Statistical analysis of CC total number/LV. Left ventricle (LV). Values are expressed as mean ± SE. a P < 0.05 vs. Control, b P < 0.05 vs. Pro + l-NAME, c P < 0.05 vs. 17β-estradiol. Images are representative of at least (AAR, n = 4; IA, n = 4; ANR, n = 3; CC, n = 6) different experiments
NO· levels and eNOS activation
NO· levels were analyzed in both the right ventricle (RV) and in the LV from all groups at the end of the protocols. In Fig. 2a is shown that changes in NO· levels followed a similar pattern in both ventricles, although changes were more evident in the LV. A 69 % diminution in NO· was observed in the Control group as compared with the Sham and the Pro groups (P < 0.05). NO· content was partially maintained in the 17β-estradiol group and totally depressed by the PI3K inhibitor LY294002 (Pro + LY group) suggesting the participation of the PI3K/Akt signaling pathway in NO· production induced by Prolame. Myocardial PHO-eNOS content correlated with variations observed in NO· levels in all groups (Fig. 2b).

Myocardial NO· levels and eNOS phosphorylation in reperfused hearts treated with Prolame. a Nitric oxide content. b eNOS phosphorylation at Ser1177 in left ventricle (LV) from Control, Pro, Pro + l-NAME, and 17β-estradiol groups. NO· content and eNOS phosphorylation in the right ventricle (RV) from the Sham group is also shown. Data are expressed as mean ± SE of six different preparations from all the experimental groups. a P < 0.05 vs. Sham, b P < 0.05 vs. Control, c P < 0.05 vs. Pro + l-NAME and 17β-estradiol
Activation of the PI3K/Akt/eNOS cascade by Prolame and its effect on NR
It has been reported that binding of estradiol to its membranal receptor activates the PI3K/Akt pathway resulting in eNOS phosphorylation; thus, we evaluated the activation of these kinases at the end of stabilization, ischemia and at 30, 60 and 90 min of reperfusion in the Control and Pro groups. There were no significant differences in PHO-PI3K content during stabilization or ischemia between Control and Pro groups. At 30 min of ischemia, levels drastically diminished in both groups (P < 0.05 vs. Stabilization). Reperfusion induced further decline in PHO-PI3K content in the Control group. On the other hand, PHO-PI3K levels increased significantly in the Pro group since the first 30 min of reperfusion and remained so, until the end of the experiment. The inhibitory effect of LY was observed in all conditions (Fig. 3a). We also correlated the effect of PI3K inhibition with IA, ANR and CC in the Pro group. Such parameters increased significantly in the Pro + LY group as compared with the Pro group (Fig. 3b). On the other hand, PHO-Akt levels were the same in the stabilization period and final ischemia in all groups. Then, at 30 min of reperfusion, levels drastically diminished and remained low throughout the 60 and 90 min of reperfusion in Control and pro groups (Fig. 4).

PI3K activation during early reperfusion is related with diminution of the no-reflow phenomenon. a Representative western blots of PHO-PI3K and PI3K content in independent preparations of LV from control, Pro and Pro + LY groups subjected to stabilization, ischemia and reperfusion for 30, 60 and 90 min. Bars represent mean ± SE of at least six independent experiments of each condition in every group. a P < 0.05 vs. stabilization. b Representative images of IA, ANR (scale 1.5 mm) and CC (scale 200 µm). A capillary compression is shown with a yellow arrow. c Statistical analyses of IA, ANR and CC. Data are expressed as mean ± SE of six independent experiments. a P < 0.05 vs. Control b P < 0.05 vs. Pro

Effect of Prolame on Akt activation in reperfused hearts. Representative western blots of Akt and PHO-Akt content from independent experiments of each condition in the control and Pro group. Bars show the mean ± SE of total and phosphorylated protein ratio from six left ventricles obtained at the indicated conditions. a P < 0.05 vs. control
PHO-eNOS levels were similar during the stabilization period and at the end of ischemia in all groups. Phosphorylation at Ser1177 increased early during reperfusion and was maintained until the end of reperfusion in the Pro group. l-NAME pretreatment (Pro + l-NAME) significantly reduced eNOS activity compared with the Pro group (P < 0.01). 17β-estradiol exerted a delayed and time-limited effect on eNOS phosphorylation around 60 min of reperfusion. Accordingly, CC in the Pro group (22 ± 3.6/LV) was significantly minor than that obtained in the Pro + l-NAME group (40 ± 2.7/LV, P < 0.05) and in the 17β-estradiol group (31 ± 4/LV, P < 0.05) (Fig. 5).

PHO-eNOS content and ANR in reperfused hearts treated with Pro, Pro + l-NAME and 17β-estradiol. a Representative western blots of PHO-eNOS content from independent preparations of LV subjected to stabilization, ischemia and reperfusion for 30, 60 and 90 min from each group (n = 6). b Transillumination images of Microfil-perfused coronary arteries from Pro, Pro + l-NAME and 17β-estradiol groups, in which capillary compressions are indicated with yellow arrows (scale 200 µm). Data are expressed as mean ± SE. a P < 0.05 vs. control, b P < 0.05 vs. Pro
Prolame stimulates NO· production in human umbilical cord vein endothelial cells (HUVEC)
In order to confirm that Prolame directly activated endothelial NO· production, studies were performed in HUVEC primary cultures. NO· levels increased significantly in the supernatants after incubation with both Prolame and with the vasoactive peptide BK, supporting the idea that the cardioprotection conferred by Prolame involved preservation of endothelium function. To further characterize the precise mechanisms underlying endothelium-dependent NO· signaling in response to Prolame, we used the estrogen receptor down-regulator (ICI) in cell cultures. HUVEC generated and maintained NO· production throughout time, peaking at 15 min (14.3 ± 2.6 µM). NO· diminished by 47.3 % in presence of the ER inhibitor (13.8 ± 2.3 vs. 7 ± 2.1 μM, P < 0.05) (Supplementary Figure 3).
Antiplatelet effect of Prolame
To unravel if the antiplatelet properties of Prolame may account in some degree for the observed cardioprotection, we measured platelet aggregation in heparinized arterial blood from control, Prolame (Pro) and (Pro + LY) rats at the end of the reperfusion experiments. Platelet aggregation observed in arterial blood from the Control group both with ADP (10 µM) and with collagen (2 µg/ml), diminished in blood samples from Pro and Pro + LY groups. Inhibition of NO production had no effect on Prolame’s antiplatelet aggregation properties (Supplementary Figure 4).
Discussion
The patency of infarct-related artery has to be restored as soon as possible to recover heart function in patients with ST-segment elevation acute myocardial infarction or acute coronary syndrome. PCI is the most direct and effective method to reperfuse the myocardium [14, 28]; however, successful reopening of the infarct-related artery (stenosis ≤10 %) not always is translated into complete tissue reperfusion; not even into obtaining TIMI grade 3 of antegrade flow. This condition, known as NR, is a complication of PCI and is closely correlated with worse prognosis, higher mortality and incidence of re-infarction in hospital [40].
Although the exact mechanism of NR is not clear, there is considerable evidence suggesting that this phenomenon is due to microvascular spasm caused by free radicals, endothelin, angiotensin-II, thromboxane and/or to progressive accumulation of leucocytes and erythrocytes in the microcirculation, to distal-end occlusion resulting from microthrombus or plaque fragments and to intracellular edema (interstitial substance). Also, mechanical obstruction by cardiac pericytes has also been pointed out as cause of diminished reperfusion of coronary capillaries [39]. Myocardial ischemia and reperfusion are responsible for a cascade of reactions leading to endothelial injury, characterized by a decrease in the production of nitric oxide (NO·); therefore, the pharmacotherapy of NR has focused primarily on strategies like local vasodilator therapy, antiplatelet therapy and use of devices for aspiration of thrombi. The use of intracoronary vasodilators has produced inconclusive results, although class IIa recommendation for the administration of intracoronary vasodilators (e.g., adenosine, calcium channel blockers, or nitroprusside) was given by the ACC/AHA PCI (American College of Cardiology/American Heart Association Percutaneous Coronary Intervention) guidelines in 2011 [31]. Two multicenter studies (AMISTAD 1 and II) showed that adenosine infusion reduced infarct size in anterior myocardial infarction [35, 41], whereas in the ADAPT trial (adenosine administration during primary percutaneous coronary intervention in acute myocardial infarction trial, n = 488), no significant differences were observed in no-reflow between patients randomized to adenosine or placebo treatment [8]. Some results have shown that nitroglycerin has little impact on arteriolar tone and hence in NR, since it needs to be metabolized in the vascular wall to release nitric oxide. Besides, only the epicardial arteries are capable to metabolize nitroglycerin but not the microvascular resistance arterioles and indeed, some studies indicate that verapamil may be more effective than nitroglycerin [49]. This calcium channel blocker, as well as diltiazem and nicardipine are endothelium-independent vasodilators used in hypertensive emergency and in NR treatment in some countries; however, it has been observed that its administration also induces bradycardia and hypotension [9]. Devices for aspiration of thrombi and thrombus-derived vasoconstrictor, have shown to reduce thrombus burden, improve perfusion, and provide protection in patients with acute myocardial infarction [22].
The estrogen receptors, being widely expressed in the cardiovascular system constitute potential therapeutic targets in acute myocardial infarction and NR [7]. Studies in vitro and in vivo have demonstrated that estrogen induces microvascular dilation by endothelium-mediated mechanisms. Activation of eNOS through non-genomic pathways, namely the PI3K/Akt cascade, was observed in human endothelial cells treated with 17β-estradiol in association with beneficial effects on the vasculature [18].
Other cardioprotective approaches like mild hypothermia and postconditioning (PostC) have been tested to reduce no-reflow. In a first report, Kloner’s group found that hypothermia protects against no-reflow but failed to reduce myocardial infarct size in rabbit hearts [16]. The same group developed a new cooling system to produce rapid hypothermia, which resulted in a profound diminution in infarct size and anatomic zone of no-reflow in hearts from rat and rabbit [19]. On the other hand, PostC, despite its almost universal efficiency in diminishing infarct size, had no effect on non-reflow in an open-chest rabbit model [17].
It is worth mentioning that both myocardial tolerance to injury from I/R and cardioprotection might be confounded by age, sex, comorbidities, and drugs, although they are relevant in patients who need cardioprotection [21]. Particularly, some intriguing results of the effects of cardioprotective strategies when antiplatelet therapy has been previously applied to patients, have raised the idea that the cardioprotective phenomenon might already be established.
Lack of conclusive reports on the efficacy of therapies against no-reflux led us to conduct this study, in which was hypothesized that Prolame, an estradiol analog with antiplatelet and antithrombotic properties may prevent NR phenomenon by exerting endothelial protection, contributing to confer cardioprotection. We found that Prolame effectively preserved cardiac function, decreased the infarcted area and the NR phenomenon in post-ischemic hearts; such protective effect was completely suppressed by inhibiting eNOS or PI3K, suggesting that this compound may be a postconditioning mimetic. The retrospective study by Roubille et al. (2012) [42] adds to this hypothesis with the demonstration that the antiplatelet compound clopidrogel attenuates lethal reperfusion injury in patients by mechanisms related with increased eNOS phosphorylation [46]. Besides, Yang et al. (2013) [51] reported that the inhibitors of the RISK pathway blocked the cardioprotection provided by clopidrogel or cangrelor in isolated rabbit hearts and also, that such results were consistent with those obtained in monkey hearts [50].
We recognize some limitations in this study: one is related with the demonstration of differential RISK pathway activation observed during postconditioning among different species, i.e., small mammals and pigs [47, 48] which raises doubts about the relevance of Akt and ERK1/2 activation in a translational context. In this regard, as it is common that even small variation in experimental strategies, not to mention the use of different species may affect cardioprotection, it would be desirable to further confirm findings obtained in rodent models, in larger animal models. Second, the experimental acute infarction used here does not reproduce the most common clinical situations of coronary occlusion resulting from chronic pathologies. However, it is worth mentioning, that no adverse effects by Prolame administration were observed in rat hearts not subjected to ischemia and reperfusion.
References
Amit G, Cafri C, Yaroslavtsev S, Fuchs S, Paltiel O, Abu-Ful A, Weinstein JM, Wolak A, Ilia R, Zahger D (2006) Intracoronary nitroprusside for the prevention of the no-reflow phenomenon after primary percutaneous coronary intervention in acute myocardial infarction. A randomized, double-blind, placebo-controlled clinical trial. Am Heart J 152(887):e9–e14. doi:10.1016/j.ahj.2006.05.010
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87:1620–1624. doi:10.1073/pnas.87.4.1620
Brady AJ, Warren JB, Poole-Wilson PA, Williams TJ, Harding SE (1993) Nitric oxide attenuates cardiac myocyte contraction. Am J Physiol 265:H176–H182
Butler MJ, ChanW Taylor AJ, Dart AM, Duffy SJ (2011) Management of the no-reflow phenomenon. Pharmacol Ther 132:72–85. doi:10.1016/j.pharmthera.2011.05.010
Coral-Vazquez RM, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP (1999) Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 98:465–474. doi:10.1016/S0092-8674(00)81975-3
Fernández-G JM, Rubio-Arroyo MF, Soriano-García M, Toscano RA, Pérez-César MC (1985) Synthesis and molecular structure of prolame, N-(3-hydroxy-1,3,5(10)-estratrien-17 beta-yl)-3-hydroxypropylamine; an amino-estrogen with prolonged anticoagulant and brief estrogenic effects. Steroids 45:151–157
Florian M, Lu Y, Angle M, Magder S (2004) Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids 69:637–645. doi:10.1016/j.steroids.2004.05.016
Fokkema ML, Vlaar PJ, Vogelzang M, Gu YL, Kampinga MA, de Smet BJ, Jessurun GA, Anthonio RL, van den Heuvel AF, Tan ES, Zijlstra F (2009) Effect of high-dose intracoronary adenosine administration during primary percutaneous coronary intervention in acute myocardial infarction: a randomized controlled trial. Circ Cardiovasc Interv 2:323–329. doi:10.1161/CIRCINTERVENTIONS.109.858977.109.858977
Fugit MD, Rubal BJ, Donovan DJJ (2000) Effects of intracoronary nicardipine, diltiazem and verapamil on coronary blood flow. Invasive Cardiol 12:80–85
Galasso G, Schiekofer S, D’Anna C, Di Gioia G, Piccolo R, Niglio T, De Rosa R, Strisciuglio T, Cirillo P, Piscione F, Trimarco B (2014) No-reflow-phenomenon: pathophysiology, diagnosis, prevention and treatment. Angiology 65:180–189. doi:10.1177/0003319712474336
Garg UC, Hassid A (1989) Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83:1774–1777. doi:10.1172/JCI114081
González G, Alvarado-Vasquez N, Fernández-G JM, Cruz-Robles D, Del Valle L, Pinzón E, Torres I, Rodriguez E, Zapata E, Gómez-Vidales V, Montaño LF, de la Peña A (2010) The antithrombotic effect of the aminoestrogenprolame (N-(3-hydroxy-1,3,5(10)-estratrien-17B-YL)-3-hydroxypropylamine) is linked to an increase in nitric oxide production by platelets and endothelial cells. Atherosclerosis 208:62–68. doi:10.1016/j.atherosclerosis.2009.06.017
Granger DL, Taintor RR, Boockvar KS, Hibbs JB Jr (1996) Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol 268:142–151
Grines CL, Westerhausen DR Jr, Grines LL, Hanlon JT, Logemann TL, Niemela M, Weaver WD, Graham M, Boura J, O’Neill WW, Balestrini C, Air PAMI Study Group (2002) A randomized trial of transfer for primary angioplasty versus on-site thrombolysis in patients with high-risk myocardial infarction: the air primary angioplasty in myocardial infarction study. J Am Coll Cardiol 39:1713–1719. doi:10.1016/S0735-1097(02)01870-3
Gross SS, Wolin MS (1995) Nitric oxide: pathophysiological mechanisms. Ann Rev Physiol 57:737–769. doi:10.1146/annurev.ph.57.030195.003513
Hale SL, Herring MJ, Kloner RA (2013) Delayed treatment with hypothermia protects against the no-reflow phenomenon despite failure to reduce infarct size. J Am Heart Assoc 2:e004234. doi:10.1161/JAHA.112.004234
Hale SL, Mehra A, Leeka J, Kloner RA (2008) Postconditioning fails to improve no reflow or alter infarct size in an open-chest rabbit model of myocardial ischemia-reperfusion. Am J Physiol Heart Circ Physiol 294:H421–H425
Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR (2003) Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem 278:2118–2123. doi:10.1074/jbc.M210828200
Herring MJ, Dai W, Hale SL, Kloner RA (2014) Rapid induction of hypothermia by the ThermoSuit system profoundly reduces infarct size and anatomic zone of no reflow following ischemia-reperfusion in rabbit and rat hearts. J Cardiovasc Pharmacol Ther (in press) pii: 1074248414535664
Heusch G (2010) Adenosine and maximum coronary vasodilation in humans: myth and misconceptions in the assessment of coronary reserve. Basic Res Cardiol 105:1–5. doi:10.1007/s00395-009-0074-7
Heusch G (2013) Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381:166–175. doi:10.1016/S0140-6736(12)60916-7
Heusch G, Kleinbongard P, Böse D, Levkau B, Haude M, Schulz R, Erbel R (2009) Coronary microembolization: from bedside to bench and back to bedside. Circulation 120:1822–1836. doi:10.1161/CIRCULATIONAHA.109.888784
Heusch G, Kleinbongard P, Skyschally A (2013) Myocardial infarction and coronary microvascular obstruction: an intimate, but complicated relationship. Basic Res Cardiol 108:380–382. doi:10.1007/s00395-013-0380-y
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 84:9265–9269. doi:10.1073/pnas.84.24.9265
Iwakura K, Ito H, Kawano S, Okamura A, Kurotobi T, Date M, Inoue K, Fujii K (2006) Chronic pre-treatment of statins is associated with the reduction of the no-reflow phenomenon in the patients with reperfused acute myocardial infarction. Eur Heart J 27:534–539. doi:10.1093/eurheartj/ehi715
Jaimez R, Cooney A, Jackson K, Lemus AE, Lemini C, Cárdenas M, García R, Silva G, Larrea F (2000) In vivo estrogen bioactivities and in vitro estrogen receptor binding and transcriptional activities of anticoagulant synthetic 17beta-aminoestrogens. J Steroid Biochem Mol Biol 73:59–66. doi:10.1016/S0960-0760(00)00053-4
Kalinowski L, Matys T, Chabielska E, Buczko W, Malinski T (2002) Angiotensin II AT1 receptor antagonists inhibit platelet adhesion and aggregation by nitric oxide release. Hypertension 40:521–527. doi:10.1161/01.HYP.0000034745.98129.EC
Keeley EC, Boura JA, Grines C (2003) Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 361:13–20. doi:10.1016/S0140-6736(03)12113-7
Kloner RA, Ganote CE, Jennings RB (1974) The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest 54:1496–1508. doi:10.1172/JCI107898
Lee CH, Wong HB, Tan HC, Zhang JJ, Teo SG, Ong HY, Low A, Sutandar A, Lim YT (2005) Impact of reversibility of no reflow phenomenon on 30-day mortality following percutaneous revascularization for acute myocardial infarction-insights from a 1,328 patient registry. J Interv Cardiol 18:261–266. doi:10.1111/j.1540-8183.2005.00041.x
Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B, Chambers CE, Ellis SG, Guyton RA, Hollenberg SM, Khot UN, Lange RA, Mauri L, Mehran R, Moussa ID, Mukherjee D, Nallamothu BK, Ting HH, ACCF, AHA, SCAI (2011) 2011 ACCF/AHA/SCAI Guideline for percutaneous coronary intervention: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 58:e44–e122. doi:10.1016/j.jacc.2011.08.007
Li XD, Yang YJ, Geng YJ, Zhang HT, Cheng YT, Wu YL (2012) Phosphorylation of endothelial NOS contributes to simvastatin protection against myocardial no-reflow and infarction in reperfused swine hearts: partially via the PKA signaling pathway. Acta Pharmacol Sin 33:879–887. doi:10.1038/aps
Loke KE, McConnell P, Tuzman JM, Shesely EG, Smith CJ, Stackpole CJ, Thompson CI, Kaley G, Wolin MS, Hintze T (1999) Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ Res 84:840–845. doi:10.1161/01.RES.84.7.840
Ma XL, Weyrich AS, Lefer DJ, Lefer AM (1993) Diminished basal nitric oxide release after myocardial ischemia and reperfusion promotes neutrophil adherence to coronary endothelium. Circ Res 72:403–412. doi:10.1161/01.RES.72.2.403
Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB (1999) Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol 34:1711–1720. doi:10.1016/S0735-1097(99)00418-0
Mandoki JJ, Zavala E, Silva G, Mendoza-Patiño N, Rubio-Póo C, Medina-Martínez S, Domínguez-Escoto P (1983) The dual effects of estrogens on blood clotting time. Proc West Pharmacol Soc 26:45–48
Murphy E (2011) Estrogen signaling and cardiovascular disease. Circ Res 109:687–696. doi:10.1161/CIRCRESAHA.110.236687
Niccoli G, Burzotta F, Galiuto L, Crea F (2009) Myocardial no-reflow in humans. J Am Coll Cardiol 54:281–292. doi:10.1016/j.jacc.2009.03.054
O´Farrell F, Attwell D (2014) A role for pericytes in coronary no-reflow. Nat Rev Cardiol 7:427–432. doi:10.1038/nrcardio.2014.58
Resnic FS, Wainstein M, Lee MK, Behrendt D, Wainstein RV, Ohno-Machado L, Kirshenbaum JM, Rogers CD, Popma JJ, Piana R (2003) No-reflow is an independent predictor of death and myocardial infarction after percutaneous coronary intervention. Am Heart J 145:42–46. doi:10.1067/mhj.2003.36
Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW, AMISTAD-II Investigators (2005) A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol 45:1775–1780. doi:10.1016/j.jacc.2005.02.061
Roubille F, Lairez O, Mewton N, Rioufol G, Ranc S, Sanchez I, Cung TT, Elbaz M, Piot C, Ovize M (2012) Cardioprotection by clopidogrel in acute ST-elevated myocardial infarction patients: a retrospective analysis. Basic Res Cardiol 107:275. doi:10.1007/s00395-012-0275-3
Roubille F, Tardif J-C (2013) Cardioprotection—time to take into account clinical complexity: the case of antiplatelet agents. Cardiovasc Drugs Ther 27:105–107. doi:10.1007/s10557-013-6443-3
Salarifar M, Mousavi MR, Saroukhani S, Nematipour E, Kassaian SE, Alidoosti M, Poorhosseini HR, Haji-Zeinali AM, Nozari Y, Hosseini K, Jalali A (2014) Percutaneous coronary intervention to treat chronic total occlusion: predictors of technical success and one-year clinical outcome. Tex Heart Inst J 41:40–47. doi:10.14503/THIJ-12-2731
Sastry KV, Moudgal RP, Mohan J, Tyaqi JS, Rao GS (2002) Spectrophotometric determination of serum nitrite and nitrate by cooper-cadmium alloy. Anal Biochem 306:79–82. doi:10.1006/abio.2002.5676
Schäfer A, Fraccarollo D, Pförtsch S, Loch E, Neuser J, Vogt C, Bauersachs J (2011) Clopidogrel improves endothelial function and NO bioavailability by sensitizing adenylyl cyclase in rats with congestive heart failure. Basic Res Cardiol 106:485–494. doi:10.1007/s00395-011-0153-4
Schwartz LM, Lagranha CJ (2006) Ischemic postconditioning during reperfusion activates Akt and ERK without protecting against lethal myocardial ischemia-reperfusion injury in pigs. Am J Physiol Heart CircPhysiol 290:H1011–H1018. doi:10.1007/s00395-014-0436-7
Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, Schulz R, Heusch G (2009) Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res 104:15–18. doi:10.1161/CIRCRESAHA.108.186429
Werner GS, Lang K, Kuehnert H, Figulla H (2002) Intracoronary verapamil for reversal of no-Reflow during coronary angioplasty for acute myocardial infarction. Catheter Cardiovasc Interv 57:444–451. doi:10.1002/ccd.10375
Yang X-M, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV (2013) Platelet P2Y12 Blockers Confer Direct Postconditioning-like Protection in Reperfused Rabbit Hearts. J Cardiovasc Pharmacol Ther 18:251–262. doi:10.1177/1074248412467692
Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV (2013) Two classes of anti-platelet drugs reduce anatomical infarct size in monkey hearts. Cardiovasc Drugs Ther 27:109–115. doi:10.1007/s10557-012-6436-7
Zhao JL, Yang YJ, Cui CJ, You SJ, Wu YJ, Gao RL (2006) Different effects of adenosine and calcium channel blockade on myocardial no-reflow after acute myocardial infarction and reperfusion. Cardiovasc Drugs Ther 20:167–175. doi:10.1007/s10557-006-8284-9
Acknowledgments
This article is part of the doctoral thesis of Sauri Hernández-Reséndiz in the Biomedical Sciences Doctoral Program, School of Medicine, National Autonomous University of Mexico (UNAM). Sauri Hernández-Reséndiz received a scholarship (32006) from the National Council of Science and Technology (CONACYT). This work was partially supported by Grant 177527 to CZ from CONACYT, Mexico.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
All authors in this work gave their informed consent prior to their inclusion in the study. The manuscript does not contain clinical studies or patient data.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hernández-Reséndiz, S., Palma-Flores, C., De los Santos, S. et al. Reduction of no-reflow and reperfusion injury with the synthetic 17β-aminoestrogen compound Prolame is associated with PI3K/Akt/eNOS signaling cascade. Basic Res Cardiol 110, 1 (2015). https://doi.org/10.1007/s00395-015-0464-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-015-0464-y