
Abstract
Acting via a cell surface receptor on integrin αvβ3, thyroid hormone is pro-angiogenic. Nongenomic mechanisms of actions of the hormone and hormone analogues at αvβ3 include modulation of activities of multiple vascular growth factor receptors and their ligands (vascular endothelial growth factor, basic fibroblast growth factor, platelet-derived growth factor, epidermal growth factor), as well as of angiogenic chemokines (CX3 family). Thyroid hormone also may increase activity of small molecules that support neovascularization (bradykinin, angiotensin II) and stimulate endothelial cell motility. Therapeutic angio-inhibition in the setting of cancer may be opposed by endogenous thyroid hormone, particularly when a single vascular growth factor is the treatment target. This may be a particular issue in management of aggressive or recurrent tumors. It is desirable to have access to chemotherapies that affect multiple steps in angiogenesis and to examine as alternatives in aggressive cancers the induction of subclinical hypothyroidism or use of antagonists of the αvβ3 thyroid hormone receptor that are under development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumor-relevant angiogenesis as a clinical target in cancer management became a reality with the work of the Folkman laboratory [1]. Metabolic differences between normal cells and cancer cells have been appreciated in terms of tolerance of hypoxia, dependence on aerobic glycolysis and preference for lowered extracellular pH in the immediate cellular microenvironment, but reduction in blood supply has been conceived to be as destructive to solid tumors as it is to normal tissues. However, the pharmacology of anti-angiogenesis in tumors that achieves clinical goals has been hampered by the complex molecular physiology of angiogenesis and redundancy of the modulators of blood vessel formation available to tumor cells [2, 3]. Initiation and maintenance of vascular supply involves local release of vascular growth factors—e.g., vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF; FGF2), platelet-derived growth factor (PDGF)—and discrete receptors for each of these proteins on the cell surface that transduce specific growth factor signals into intracellular and extracellular angiogenesis-related events. Targeting of one or two of these growth factors or receptors pharmacologically does leave other vascular growth factor options to cancer cells and these include epidermal growth factor (EGF) [4, 5] and insulin-like growth factor-1 (IGF-1) [6–8], in addition to the factors listed above. Function of the growth factor receptors may be modulated by adjacent plasma membrane integrins, such as αvβ3 [9–12], a structural protein of the plasma membrane [13].
Nonpeptide hormones may also support angiogenesis. Estrogen and progesterone are pro-angiogenic in their normal target tissues and estrogen may be a factor in tumor-relevant blood vessel formation in nuclear estrogen receptor (ER)-bearing cancers, such as lung [14, 15]. It is now clear that thyroid hormone is pro-angiogenic via its cell surface receptor on integrin αvβ3. Because this integrin is expressed generously by cancer cells and rapidly dividing blood vessel cells, thyroid hormone (l-thyroxine, T4; 3, 5, 3′-triiodo-l-thyronine, T3) may be an important contributor to blood supply maintenance about cancers. This pro-angiogenic activity may be blocked by tetraiodothyroacetic acid (tetrac), the deaminated metabolite of T4, which is not, itself, pro-angiogenic, but blocks binding of T4 and T3 at the hormone receptor site on αvβ3 [16]. Tetrac and its nanoparticulate formulation have also been shown in the absence of T4 and T3 to act via the integrin to inhibit activity of multiple vascular growth factors. These actions of tetrac and nanoparticulate tetrac are reviewed below.
There are other strategies to consider as points of attack in the vascular infrastructure of solid tumors. Vascular microtubule formation is a function of endothelial cell motility and cell-to-cell adhesion. Interference with these endothelial cell functions in the environment of tumors would be therapeutically desirable. Neovascularization around the tumor requires that the intercellular matrix structure of the existing vascular bed be loosened to permit vascular budding and vessel formation. Factors such as angiopoietin-2 (Ang-2) are premonitory contributors to such changes in vascular beds and when paired with VEGF promote angiogenesis [17]. Thrombospondin 1 (TSP1) gene expression is almost invariably suppressed in cancer cells; the gene product is an endogenous anti-angiogenic factor [18]. Relief of suppression of transcription of TSP1 is desirable in chemotherapy. While it is feasible to block vascular growth receptor function with monoclonal antibody to specific receptors or by interference with crosstalk between integrin αvβ3 and vascular growth factor receptors clustered with the integrin, it would also be desirable to decrease specific vascular growth factor receptor gene expression or local cancer cell release of just-synthesized protein growth factors. Partially successful anti-angiogenesis selects for hypoxia-tolerant tumor cells that are radioresistant. Radioresistance requires an intact cellular mechanism for rapid repair of double-strand DNA breaks induced by radiation; it would be desirable for the anti-angiogenic process to include suppression of radiation defense mechanisms that depend, for example, on cancer cell expression of genes such as hypoxia-inducible factor-1α (HIF-1α) [19]. HIF-1α protein is a nuclear transactivator whose activity is also relevant to VEGF gene expression and other important tumor cell functions [20].
A few inhibitors of vascular growth factors or their receptors that are currently available do interfere with the actions of more than one growth factor or angiogenesis-relevant protein. Ziv-aflibercept complexes with VEGF-A/VEGF-B and placental growth factor (PlGF) [21] and tyrosine kinase inhibitors such as sunitinib affect activities of both VEGF and PDGF [22]. It has seemed unreasonable, however, to expect to develop anti-angiogenic agents for use against cancer that affect the full complement of vascular growth factor defenses mentioned above that are available to blood vessels and tumor cells co-engaged in angiogenesis.
However, the appreciation of the existence of crosstalk between αvβ3 integrin and multiple growth factor receptors—either physically via their extracellular domains or chemically at the level of the plasma membrane or signal transduction systems available to the endofacial surface of the plasma membrane—has encouraged a search for αvβ3-targeted agents that are anti-angiogenic. A specialized domain of the integrin designated the Arg-Gly-Asp (RGD) recognition site can be disordered with cyclic RGD peptides and affect integrin-vascular growth factor receptor communication. In this review, however, we concentrate on a small molecule receptor for thyroid hormone analogues on αvβ3 and links of this receptor to angiogenesis. At this receptor, thyroid hormone analogues relate to transcription of a spectrum of angiogenesis-relevant genes, as well as to crosstalk between the integrin and receptors for at least five vascular growth factors (see next section).
Thyroid hormone and hormone analogues on actions of vascular growth factor cytokines
The mechanism of thyroid hormone-induced neovascularization was described in part a decade ago in the chick chorioallantoic membrane (CAM) assay of angiogenesis [23, 24]. Initiated at the hormone receptor on αvβ3 in blood vessel cells, the effect was shown to depend at least in part upon the transcription of basic fibroblast growth factor (bFGF; FGF2) gene and increased cell release of bFGF with autocrine stimulation of angiogenesis. Unmodified tetrac blocked this agonist action of T4 and T3. We subsequently showed that, in the absence of agonist thyroid hormone, tetrac blocked the activity of VEGF and bFGF added to the CAM assay [25]. It was proposed that disordering of crosstalk between the integrin and VEGFR and bFGFR explained this effect of tetrac. However, VEGF gene expression is also regulated by HIF-1α [26], a transcription factor whose abundance in cells is regulated by thyroid hormone via αvβ3 [27]. We have also shown that PDGF is inactive in the CAM assay in the presence of tetrac (SA Mousa: unpublished). We now know that function of the IGF1R is modulated by thyroid hormone analogues [28], as are activities of the EGFR [29] and transcription of the EGFR gene [30]. Our survey of plasma membrane proteins extracted from a variety of cells for binding of radiolabeled thyroid hormone has disclosed binding only by αvβ3 [24].
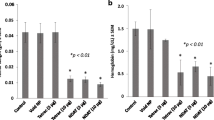
Studies of the effects in the CAM assay of a nanoparticulate formulation of tetrac that restricts tetrac to the extracellular space and αvβ3 have confirmed its anti-angiogenic activities. In this formulation, tetrac is covalently bound through a linker to the surface of a 200 nm nanoparticle that precludes cell entry [31]. Thus, a component of the anti-angiogenic properties of Nanotetrac and tetrac relate to integrin-dependent actions on functions of multiple vascular growth factors and of growth factor receptors. Actions of T4, nanoparticulate T4 and Nanotetrac on angiogenesis in the CAM are shown in Fig. 1.

Angiogenic activities of thyroid hormone (T4), nanoparticulate T4 (as T4-agarose, T4-ag) and tetrac in the chick chorioallantoic membrane (CAM) model. CAM assay methodology is as we have previously described [23]. Nanoparticulate T4 cannot enter the cell. The pro-angiogenic actions of T4 and T4-ag are similar in magnitude, indicating initiation of the effects at integrin αvβ3 on the cell surface. Tetrac eliminates the vascular activities of T4 and T4-ag
Thyroid hormone analogues and endogenous small pro-angiogenic molecules
We have recently examined the pro-angiogenic activities of bradykinin, angiotensin II (Ang II) and lipopolysccharide (LPS) in the CAM and their responses to Nanotetrac (NT) (Table 1) (see Discussion). Quantitation of vascular branch points in the CAM permits comparison of effectiveness of different agents or of concentrations of single agents. In the case of each of the substances included in the Table, the pro-angiogenic properties were markedly diminished by Nanotetrac. The ability of Nanotetrac to oppose the pro-angiogenic activity of VEGF and bFGF confirmed a previous report [25]. The combination of VEGF, bFGF and tumor necrosis factor-α (TNF-α) was also examined and exhibited the most vigorous neovascularization in this set of experiments. This activity of the combination was inhibited by Nanotetrac.
It should also be noted that T3 was more potent than T4 in the assay (Table 1). However, T3 will not contribute materially to angiogenesis when the nonthyroidal illness (NTI) syndrome has complicated clinical cancer and circulating endogenous T3 levels are low [32].
Thyroid hormone analogues and TSP1
A survey of the actions of unmodified tetrac and Nanotetrac on gene expression in breast cancer cells revealed that basally suppressed TSP1 expression in such cells was reversed by these hormone analogues [30]. This is a desirable quality in an agent with other anti-angiogenic properties.
Thyroid hormone analogues and cell motility
Endothelial cell motility is essential to new blood vessel formation. We have examined human endothelial cell motility in a modified Boyden chamber and have found that T4 and T3 (each at 0.1 μM total concentration) increase migration rate of the cells towards a chemical cue (vitronectin) by more than twofold and threefold, respectively (Fig. 2). The T4 concentration yields a physiologic free level of hormone, whereas the T3 level is supraphysiologic. Tetrac (1 μM) blocks cell migration induced by T4 and T3. Integrity of the cytoskeleton is essential to efficient motility and thyroid hormone, specifically T4, has been shown by Farwell and Leonard to support the conversion of soluble actin to F-actin [33]. This activity was described prior to the description of the iodothyronine receptor on integrin αvβ3, but the integrin has been implicated in control of the actin cytoskeleton [34].

Effects of T4 and T3, with and without unmodified tetrac, on human dermal endothelial cell (EC) migration towards a vitronectin (VN) cue. Cell migration assay was by our previously reported method, using a modified Boyden chamber apparatus [55]. Cells were placed in the upper chamber and VN was located in the lower chamber. Relative fluorescence units (RFU) track accumulation of cells in the lower chamber. Unmodified tetrac inhibits the actions of agonist T4 and T3 on cell migration. Standard error bars represent the means of duplicate studies carried out X3
Thyroid hormone analogues and vascular microtubule formation and budding
Primordial blood vessel formation involves vascular microtubule formation. Thyroid hormone stimulates microtubule formation in an in vitro model of human endothelial cells [35]. Vascular budding is also demonstrable in ischemic rabbit hindlimb vessels when these vessels are perfused with T4 [36]. Such studies have not specifically been extended to the tumor vascular microenvironment, but these hormone-dependent activities are assumed to occur there.
Thyroid hormone and angiogenesis-relevant chemokines
The pro-angiogenic chemokine ligands CXCL2 and CXCL3 [37] are regulated from integrin αvβ3 by thyroid hormone analogues (GV Glinsky: unpublished). The CX3C chemokine family consists of the CX3CL1/fractalkine ligand and its receptor, CX3CR1, and the genes for these proteins are also differentially regulated by thyroid hormone analogues from the hormone receptor on the integrin [38]. The CX3C axis is relevant to angiogenesis via multiple pathways that ensure maturation and structural integrity of newly-formed microvessels [37]. In the absence of this chemokine ligand and its receptor, neovascularization results in undersized, leaking vessels [39]. Nanotetrac decreases transcription of CX 3 CL1 and CX 3 CR1 in cancer cells [38].
Thyroid hormone analogues and angiogenesis in situ in tumor xenografts
Pro-angiogenic properties of thyroid hormone demonstrated in model systems such as the CAM have also been studied with formulations of the anti-angiogenic thyroid hormone analogue, tetrac, in a variety of human tumor xenografts. The latter include lung [40] and pancreas [41] cancers, renal cell carcinoma (RCC) [42], medullary thyroid carcinoma [43] and follicular thyroid cancer [44]. In all such xenografts, unmodified tetrac and nanoparticulate tetrac have rapidly decreased tumor-related blood vessel formation. Tetrac formulations are anti-angiogenic by several mechanisms, as indicated above, but a critical action of these compounds is antagonism of the pro-angiogenic activity of thyroid hormone agonists, T4 and T3, at the plasma membrane hormone receptor site on integrin αvβ3 [16].
Tumor cell proliferation directed by thyroid hormone that may be relevant to effectiveness of specific anti-angiogenesis therapy
In the presence of physiologic concentrations of thyroid hormone, there is proliferation of a variety of tumor cell lines we have studied [16]. In the context of observations we have reviewed above, pharmacological angioinhibitory cancer strategies directed at single vascular growth factors appear handicapped. That is, monoclonal antibody to VEGF, alone, or TKI treatment that reduces effectiveness only of VEGF and bFGF must work clinically in a pro-angiogenic tumor environment supported by endogenous factors such as thyroid hormone. The latter supports most of the vascular growth factor cytokine axes by several mechanisms, stimulates multiple pro-angiogenic chemokines, increases endothelial cell motility and stabilizes the cytoskeleton of motile and adherent cells. These hormonal effects may be sufficient in the setting of aggressive and/or recurrent tumors to oppose specific anti-angiogenic therapy.
Discussion
It is clear that multiple components of neovascularization are supported by thyroid hormone. In the setting of cancer chemotherapy that is primarily angioinhibitory and directed at one or no more than several vascular growth factor systems—growth factors, themselves, or their receptors—endogenous thyroid hormone is proposed by us to be a confounding influence. That is, actions of the hormone may contribute to diminished drug effectiveness in certain patients whose solid tumors are subjected to anti-angiogenesis therapy. Such patients may be those with circulating free thyroxine (FT4) levels that are in the upper quartile of the reference range or are elevated in the NTI state [32]. Patients with suppressed serum thyrotropin (TSH) levels that are consistent with NTI or with subclinical hyperthyroidism may have blood FT4 concentrations within the reference range that are interpreted by at least one organ, the pituitary gland, as elevated. We propose that such free hormone concentrations are sufficient in certain patients to be multifactorially pro-angiogenic in tumor-relevant vasculature, regardless of the presence of anti-angiogenic agents directed at one or two vascular growth factors. There is some clinical evidence to support this proposition. For example, highly vascular glioblastoma multiforme (GBM) has been shown to respond favorably to induction of mild (‘subclinical’) hypothyroidism [45]. In this GBM study, patients had already exhausted standard therapeutic measures. The vascularity of RCC has encouraged the use of anti-angiogenic therapy in this condition [46]. Interestingly, trials of TKI therapy in RCC patients has shown promise particularly when TKI-induced hypothyroidism has complicated management [47–49]. This raises the possibility that loss of thyroid hormone support for tumor cell and blood vessel cell proliferation is important to management of this condition.
Nanoparticulate tetrac is an antagonist of actions of thyroid hormone at the hormone receptor on αvβ3. The agent has been shown experimentally to have multiple anti-angiogenic actions [16], but has not undergone clinical trial. Nanotetrac inhibits binding of T4 and T3 to αvβ3, but has been shown to have a variety of anti-angiogenic properties in the absence of T4 and T3. That is, it blocks expression of pro-angiogenic chemokine CX 3 C ligand and receptor [38], stimulates expression of anti-angiogenic TSP1 gene that is almost invariably suppressed in cancer cells [30] and inhibits expression of EGFR [30]. As noted above, Nanotetrac disorders the crosstalk between αvβ3 and nearby receptors for VEGF, bFGF and PDGF. Endogenous substances in addition to vascular growth factors that are pro-angiogenic in the tumor microenvironment include Ang-II [50] and bradykinin [51]. The angiogenic activity of these agents is inhibited by Nanotetrac (Table 1). Neovascularization that is induced by LPS is also blocked by Nanotetrac. The pro-angiogenic effect of LPS is relevant to inflammation [52] and inflammation-associated cancer [53, 54].
The preclinical evidence is extensive that thyroid hormone is pro-angiogenic by a variety of mechanisms. We postulate that in some cancer patients in whom specific anti-angiogenic therapy has been ineffective, the angiogenic activity of endogenous thyroid hormone contributes to suboptimal cancer response. As noted above, spontaneous or induced hypothyroidism can favorably change the clinical courses of GBM, breast and RCC. These clinical observations may reflect proliferative effects of thyroid hormone on tumor cells, themselves [16]. However, when we have examined the vascularity of human cancer xenografts, the prompt decrease in tumor volume with anti-thyroid hormone (Nanotetrac) action at integrin αvβ3 has been associated with a 50–60 % decrease in tumor vascularity [40–44]. This is consistent with, but does not establish, the concept that endogenous thyroid hormone may be acting at αvβ3 in the host animals, in concert with tumor secretion of vascular growth factors, to support angiogenesis. However, αvβ3 is expressed generously by dividing blood vessel cells and tumor cells [16], as stated above, and the integrin is not prominent in quiescent endothelial cells in nonmalignant, normal tissues. Vascular scanning of tumors in patients in whom subclinical hypothyroidism has been medically induced—for example, with TKI therapy—will be an initial test of the contribution of thyroid hormone to tumor vascularity, when compared with euthyroid TKI-treated patients.
References
Folkman J (2006) Angiogenesis. Annu Rev Med 57:1–18
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 8:210–221
Eklund L, Bry M, Alitalo K (2013) Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol Oncol 7:259–282
Sun QM, Miao ZH, Lin LP, Gui M, Zhu CH, Xie H, Duan WH, Ding J (2009) BB, a new EGFR inhibitor, exhibits prominent anti-angiogenesis and antitumor activities. Cancer Biol Ther 8:1640–1647
Bertrand-Duchesne MP, Grenier D, Gagnon G (2010) Epidermal growth factor released from platelet-rich plasma promotes endothelial cell proliferation in vitro. J Periodontal Res 45:87–93
Ozkan EE (2011) Plasma and tissue insulin-like growth factor-1 receptor (IGF-1R) as a prognostic marker for prostate cancer and anti-IGF-1R agents as novel therapeutic strategy for refractory cases: a review. Mol Cell Endocrinol 344:1–24
Haleagrahara N, Chakravarthi S, Mathews L (2011) Insulin like growth factor-1 (IGF-1) causes overproduction of IL-8, an angiogenic cytokine, and stimulates neovascularization in isoproterenol-induced myocardial infarction in rats. Int J Mol Sci 12:8562–8574
Piecewicz SM, Pandey A, Roy B, Xiang SH, Zetter BR, Sengupta S (2012) Insulin-like growth factors promote vasculogenesis in embryonic stem cells. PLoS One 7(2):e32191
Schneller M, Vuori K, Ruoslahti E (1997) Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J 16:5600–5607
Tsou R, Isik FF (2001) Integrin activation is required for VEGF and FGF receptor protein presence on human microvascular endothelial cells. Mol Cell Biochem 224:81–89
De S, Razornova O, McCabe NP, O’Toole T, Qin J, Byzova TV (2005) VEGF-integrin interplay controls tumor growth and vascularization. Proc Natl Acad Sci USA 102:7589–7594
Montenegro CF, Salla-Pontes CL, Ribeiro JU, Machado AZ, Ramos RF, Figueiredo CC, Morandi V, Selistre-de-Araujo HS (2012) Blocking αvβ3 integrin by a recombinant RGD disintegrin impairs VEGF signaling in endothelial cells. Biochimie 94:1812–1820
Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. J Biol Chem 275:21785–21788
Wei S, Said-Al-Naief N, Hameed O (2009) Estrogen and progesterone receptor expression is not always specific for mammary and gynecologic carcinomas: a tissue microarray and pooled literature review study. Appl Immunohistochem Mol Morphol 17:393–402
Shimizu K, Hirami Y, Saisho S, Yukawa T, Maeda A, Yasuda K, Nakata M (2012) Membrane-bound estrogen receptor-α expression and epidermal growth factor receptor mutation are associated with a poor prognosis in lung adenocarcinoma patients. World J Surg Oncol 10:141
Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY (2011) Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol 51:99–115
Fagiani E, Christofori G (2013) Angiopoietins in angiogenesis. Cancer Lett 328:18–26
Lawler PR, Lawler J (2012) Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med 2(5):a006627
Patiar S, Harris AL (2006) Role of hypoxia-inducible factor-1 alpha as a cancer therapy target. Endocr Relat Cancer 13(Suppl 1):S61–S75
Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, Rodriguez-Enriquez S, Moreno-Sanchez R (2009) Hif-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9:1084–1101
Wilson PM, LaBonte MJ, Lenz HJ (2013) Assessing the in vivo efficacy of biological antiangiogenic therapies. Cancer Chemother Pharmacol 71:1–12
Majumder S, Piquet AC, Dufour JF, Chatterjee S (2013) Study of the cellular mechanism of Sunitinib mediated inactivation of activated hepatic stellate cells and its implications in angiogenesis. Eur J Pharmacol 705:86–95
Davis FB, Mousa SA, O’Connor L, Mohamed S, Lin HY, Cao HJ, Davis PJ (2004) Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ Res 94:1500–1506
Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ (2005) Integrin alphavbeta 3 contains a cell surface receptor for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 146:2864–2871
Mousa SA, Bergh JJ, Dier E, Rebbaa A, O’Connor LJ, Yalcin M, Aljada A, Dyskin E, Davis FB, Lin HY, Davis PJ (2008) Tetraiodothyroacetic acid, a small molecule integrin ligand, blocks angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor. Angiogenesis 11:183–190
Wong VW, Crawford JD (2013) Vasculogenic cytokines in wound healing. Biomed Res Int 2013:190486
Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ (2009) l-Thyroxine vs. 3,5,3′-triiodo-l-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol 296:C980–C991
Incerpi S, Lin HY, De Vito P, Fiore AM, Ahmed RG, Salvia R, Candelotti E, Luly P, Pedersen JZ, Davis FB, Davis PJ (2013) Thyroid hormone inhibition in L6 myoblasts of IGF-1-mediated glucose uptake and proliferation: new roles for integrin αvβ3. Manuscript submitted
Shih A, Zhang S, Cao HJ, Tang HY, Davis FB, Davis PJ (2004) Disparate effects of thyroid hormone on actions of epidermal growth factor and transforming growth factor-α are mediated by 3′,5′-cyclic adenosine monophosphate-dependent protein kinase II. Endocrinology 145:1708–1717
Glinskii AB, Glinsky GV, Lin HY, Tang HY, Sun M, Davis FB, Luidens MK, Mousa SA, Hercbergs AH, Davis PJ (2009) Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 8:3554–3562
Bharali DJ, Yalcin M, Davis PJ, Mousa SA (2013) Tetraiodothyroacetic acid (tetrac) conjugated PLGA nanoparticles: a nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine 8:1943–1954
Farwell AP (2013) Nonthyroidal illness syndrome. Thyroid 20:478–484
Leonard JL, Farwell AP (1997) Thyroid hormone-regulated actin polymerization in brain. Thyroid 7:147–151
Stefansson S, Su EJ, Ishigami S, Cale JM, Gao Y, Gorlatova N, Lawrence DA (2007) The contributions of integrin affinity and integrin-cytoskeletal engagement in endothelial and smooth muscle cell adhesion to vitronectin. J Biol Chem 282:15679–15689
Mousa SA, Davis FB, Mohamed S, Davis PJ, Feng X (2006) Pro-angiogenesis action of thyroid hormone and analogs in a three-dimensional in vitro microvascular endothelial sprouting model. Int Angiol 25:407–413
El-Eter E, Rebbaa H, Alkayali A, Mousa SA (2007) Role of thyroid hormone analogues in angiogenesis and the development of collaterals in the rabbit hind limb ischemia model. J Thromb Thrombolysis 5(Suppl 1):375
Owen JL, Mohamadzadeh M (2013) Macrophages and chemokines as mediators of angiogenesis. Front Physiol 4:159
Davis PJ, Glinsky GV, Lin HY, Incerpi S, Davis FB, Mousa SA, Tang HY, Hercbergs A, Luidens MK (2013) Molecular mechanisms of actions of formulations of the thyroid hormone analogue, tetrac, on the inflammatory response. Endocr Res 38:112–118
Kumar AH, Martin K, Turner EC, Buneker CK, Dorgham K, Deterre P, Caplice NM (2013) Role of CX3CR1 receptor in monocyte/macrophage driven neovascularization. PLoS One 8(2):e57230
Mousa SA, Yalcin M, Bharali DJ, Meng R, Tang HY, Lin HY, Davis FB, Davis PJ (2012) Tetraiodothyroacetic acid and its nanoformulation inhibit thyroid hormone stimulation of non-small cell lung cancer cells in vitro and their growth in xenografts. Lung Cancer 76:39–45
Yalcin M, Lin HY, Sudha T, Bharali DJ, Meng R, Tang HY, Davis FB, Stain SC, Davis PJ, Mousa SA (2013) Response of human pancreatic cancer cell xenografts to tetraiodothyroacetic acid nanoparticles. Horm Cancer 4:176–185
Yalcin M, Bharali DJ, Lansing L, Dyskin E, Mousa SS, Hercbergs A, Davis FB, Davis PJ, Mousa SA (2009) Tetraiodothyroacetic acid (tetrac) and tetrac nanoparticles inhibit growth of human renal cell carcinoma xenografts. Anticancer Res 29:3825–3831
Yalcin M, Dyskin E, Lansing L, Bharadi DJ, Mousa SS, Bridoux A, Hercbergs AH, Lin HY, Davis FB, Glinsky GV, Glinskii AB, Ma J, Davis PJ, Mousa SA (2010) Tetraiodothyroacetic acid (tetrac) and nanoparticulate tetrac arrest growth of medullary carcinoma of the thyroid. J Clin Endocrinol Metab 95:1972–1980
Yalcin M, Bharali DJ, Dyskin E, Dier E, Lansing L, Mousa SS, Davis FB, Davis PJ, Mousa SA (2010) Tetraiodothyroacetic acid and tetraiodothyroacetic acid nanoparticle effectively inhibit the growth of human follicular thyroid cell carcinoma. Thyroid 20:281–286
Hercbergs AA, Goyal LK, Suh JH, Lee S, Reddy CA, Cohen BH, Stevens GH, Reddy SK, Peereboom DM, Elson PJ, Gupta MK, Barnett GH (2003) Propylthiouracil-induced chemical hypothyroidism with high-dose tamoxifen prolongs survival in recurrent high grade glioma: a phase I/II trial. Anticancer Res 23:617–626
Aziz SA, Sznol J, Adeniran A, Colberg JW, Camp RL, Kluger HM (2013) Vascularity of primary and metastatic renal cell carcinoma specimens. J Transl Med 11:15
Hercbergs AH, Ashur-Fabian O, Garfield D (2011) Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Current Opin Endocrinol Diabetes Obes 17:432–436
Schmidinger M, Vogl UM, Bojic M, Lamm W, Heinzl H, Haitel A, Clodi M, Kramer G, Zielinski CC (2011) Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer 117:534–544
Riesenbeck LM, Bierer S, Hoffmeister J, Kopke T, Papavasilis P, Hertle L, Thielen B, Hermann E (2011) Hypothyroidism correlates with a better prognosis in metastatic renal carcinoma patients treated with sorafenib or sunitinib. World J Urol 29:807–813
Okwan-Duodo D, Landry J, Shen XZ, Diaz R (2013) Angiotensin-converting enzyme and the tumor microenvironment: mechanisms beyond angiogenesis. Am J Physiol Regul Integr Comp Physiol 305:R205–R215
Stewart JM, Gera L, Chan DC, York EJ, Simkeviciene V, Bunn PA Jr, Taraseviciene-Stewart L (2005) Combination cancer chemotherapy with one compound: pluripotent bradykinin antagonists. Peptides 26:1288–1291
Song X, Chen Y, Sun Y, Lin B, Qin Y, Hui H, Li Z, You Q, Lu N, Guo Q (2012) Oroxylin A, a classical natural product, shows a novel inhibitory effect on angiogenesis induced by lipopolysaccharide. Pharmacol Rep 64:1189–1199
Rakhesh M, Cate M, Vijay R, Shrikant A, Shanjana A (2012) A TLR4-interacting peptide inhibits lipopolysaccharide-stimulated inflammatory responses, migration and invasion of colon cancer SW480 cells. Oncoimmunology 1:1495–1506
Melkamu T, Qian X, Upadhyaya P, O’Sullivan MG, Kassie F (2013) Lipopolysaccharide enhances mouse lung tumorigenesis: a model for inflammation-driven cancer. Vet Pathol 50:895–902
Mousa SA, Mohamed S, Wexler EJ, Kerr JS (2005) Antiangiogenesis and anticancer efficiency of TA138, a novel alphavbeta3 antagonist. Anticancer Res 25:197–206
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mousa, S.A., Lin, HY., Tang, H.Y. et al. Modulation of angiogenesis by thyroid hormone and hormone analogues: implications for cancer management. Angiogenesis 17, 463–469 (2014). https://doi.org/10.1007/s10456-014-9418-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-014-9418-5