
Abstract
Angiogenesis-targeting agents, predominantly inhibitors of vascular endothelial growth factor and its receptors, have become a mainstay in oncology practice over the last decade. The approved drugs, which include two antibodies and seven small molecule inhibitors, represent strong validation for the field of anti-angiogenesis in the treatment of cancer. In addition to ongoing clinical studies assessing new indications for these agents, novel inhibitors are undergoing clinical evaluations and combination therapies of anti-angiogenic drugs with other targeted approaches are being investigated. This chapter will review the development of the currently marketed drugs targeting VEGFR, as well as the new inhibitors currently being assessed in the clinic.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Angiogenesis-targeting agents have become a mainstay of oncology practice over the last decade. The first agent was bevacizumab (Avastin®), a monoclonal antibody to Vascular Endothelial Growth Factor (VEGF), which was approved initially by the FDA in 2004 for the treatment of colorectal cancer and expanding to include approvals in certain types of lung, kidney, brain, ovarian, and cervical cancers [1]. This was followed in the following decade by small molecule inhibitors of the VEGF-receptor (VEGFR) pathway: sorafanib (Nexavar®), sunitinib (Sutent®), pazopanib (Votrient®), axitinib (Inlyta®) regorafenib (Stivarga®), nintedanib (Ofev®/Vargatef®), and lenvatinib (Lenvima®), and the VEGFR-2 antibody ramucirumab [1]. Taken together these drugs treat multiple oncology indications and represent strong validation for the field of anti-angiogenesis in the treatment of cancer that had begun with the pioneering work of Judah Folkman in 1971 [2]. The approved VEGFR inhibitors are being studied in the clinic for new indications, novel inhibitors are undergoing clinical assessment, and combination therapies of anti-angiogenic drugs with other targeted approaches are being investigated. This chapter will review the history and development of the currently marketed drugs targeting VEGFR, as well as the new inhibitors currently being assessed in the clinic.
2 Mechanism of Action
The growth of solid tumors depends on the supply of nutrients and oxygen from newly formed capillaries sprouting from existing blood vessels, a process known as angiogenesis [2]. Tumors induce angiogenesis by secretion of a number of endogenous proteins, notably VEGF, which binds to one of three transmembrane tyrosine kinase receptors, VEGFR-1,2,3, on nearby endothelial cells [3]. The VEGFRs have an extracellular portion consisting of seven immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain [4]. In response to ligand binding, the VEGFRs undergo dimerization as both homo- and hetero-dimers, facilitating the trans autophosphorylation of intracellular tyrosine kinases and subsequent activation of downstream signaling pathways that elicit various angiogenic responses (Fig. 1).


The binding mode of the ligand VEGF and receptor VEGFR-2 located on endothelial cells. In the first step, VEGF binds to domains 2 and 3 of the 7 immunoglobulin homology domains in the extracellular domain of VEGFR-2 monomer. Binding of VEGF induces the dimerization of VEGFR-2, leading to trans autophosphorylation of tyrosine sites in the intracellular kinase domain region. These provide docking sites for adaptor proteins, which initiate pro-angiogenic signal transduction cascades, resulting in the increased cellular proliferation, migration, permeability, and survival of the endothelial cells
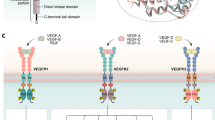
VEGFs produced by cells are 34–46-kDa homodimeric glycoproteins that act as potent mitogens that stimulate the growth of new blood vessels [3]. The mammalian VEGF gene family consists of five groups, VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PLGF (placenta growth factor), along with two structurally related proteins, VEGF-E from parapoxvirus and VEGF-F from snake venom. The VEGFRs have three main subtypes: VEGFR-1 (also known as Flt-1), VEGFR-2 (also known as KDR/Flk-1), and VEGFR-3 (also known as Flt-4). The VEGF ligands have distinctive binding specificities for the three transmembrane tyrosine kinase receptors as shown in Fig. 2 [5]. VEGF-A and VEGF-E bind to VEGFR-2, which is located on the surface of vascular endothelial cells, and is the major mediator of the mitogenic, angiogenic, and permeability enhancing effects of VEGF. VEGF-A, VEGF-B, and PLGF bind to VEGFR-1 expressed on hematopoietic stem cells, macrophages, and monocytes as well as on the vascular endothelium. The function of VEGFR-1 is less well defined, being required for recruitment of hematopoietic precursors, but also playing a negative regulatory role in embryonic angiogenesis by acting as a decoy receptor to VEGFR-2. VEGF-C and VEGF-D bind to VEGFR-3, which is largely located at the lymphatic endothelium.


Activation of the VEGFR homo- and hetero-dimer receptors by VEGF ligands
3 First Generation Marketed Anti-Angiogenesis Inhibitors in Oncology
The clinical use of anti-angiogenesis agents in oncology began with the approval of the VEGF monoclonal antibody bevacizumab in 2004 by the FDA for the treatment of colorectal cancer, followed by additional approvals in certain types of lung, kidney, brain, ovarian, and cervical cancers [1]. This first generation of anti-angiogenesis agent was followed in the following 5 years by the small molecule inhibitors of VEGFR, sorafanib, sunitinib, and pazopanib, as shown in Table 1. All three were initially approved for kidney cancer, with sunitinib receiving simultaneous approval for gastrointestinal stromal tumors, and each have subsequently received approval for additional indications; sorafanib for liver and thyroid cancers, sunitinib for pancreatic tumors, and pazopanib for soft tissue sarcomas. It should also be noted that bevacizumab was also initially approved for breast cancer, but that approval was withdrawn by the FDA when later studies showed lack of evidence of safety and efficacy for that use [1].
4 Bevacizumab (Avastin®)
As a VEGF antibody, bevacizumab strictly falls outside the scope of this chapter, but its importance as the first approved anti-angiogenesis agent and its widespread clinical use merits a brief description of its discovery and clinical development, to set the stage for the discussion on VEGFR inhibitors. The path to bevacizumab began with the discovery by Ferrara et al. at Genentech in 1989 of the VEGF growth factor isolated from medium conditioned by bovine pituitary follicular cells [6, 7]. By the early 1990s, they were able to demonstrate that a murine anti-human monoclonal antibody (MAb) to VEGF was able to potently suppress angiogenesis and growth in a variety of human tumor cells lines transplanted in nude mice, with no direct effect on tumor cells. In 1997 the same group reported that a recombinant humanized anti-VEGF MAb was able to inhibit VEGF-induced proliferation of endothelial cells in vitro and tumor growth in vivo with potency and efficacy very similar to those of murine VEGF. This humanized anti-VEGF monoclonal antibody binds selectively to VEGF-A isoforms, with a Kd ~ 0.5 nM, but does not neutralize the other members of the VEGF gene family. The human portion (accounting for 93%) contains the antibody framework and the murine portion (accounting for 7%) contains the regions that bind VEGF-A with high affinity.
Phase I clinical trials were initiated in 1997 in patients at dose levels from 0.1 to 10 mg/kg [8]. Bevacizumab showed a linear pharmacokinetic profile administered intravenously over 90 min on days 0, 28, 35, and 42 and a terminal half life of 21 days. One patient with renal cell cancer (RCC) achieved a minor response and nearly half (48%) of the remaining patients achieved disease stabilization. A phase Ib trial then assessed bevacizumab at 3 mg/kg weekly for 8 weeks in combination with a number of standard cytotoxics [9]. This showed that the antibody was relatively non-toxic and when combined with chemotherapy did not significantly exacerbate the toxicological profile. Subsequently five parallel phase II trials were initiated, three of bevacizumab alone in prostate, metastatic breast, and RCC, and two in combination with standard therapy in non-small cell lung cancer (NSCLC) and metastatic colorectal (mCRC) cancers [7]. The most encouraging results were observed when used in patients with RCC, NSCLC, and mCRC cancers [10]. In renal cancer, patients received bevacizumab 3 mg/kg or 10 mg/kg or placebo administered every 2 weeks. The percentage of patients being progression-free was 64% in the high dose group, compared to 34% and 20% in the low dose and placebo groups, respectively. Toxicity included increased blood pressure and asymptomatic proteinuria. In the NSCLC phase II trial, patients received carboplatin and paclitaxel, plus or minus bevacizumab, at 7.5 or 15 mg/kg every 3 weeks. The trial indicated no significant increase in toxicity for bevacizumab compared to chemotherapy alone, and the 15 mg/kg dose showed an increase in time to progression (7.4 versus 4.2 months) and a modest increase in survival (17.7 versus 14.9 months). In the first phase II trial in mCRC, patients received 5-fluorouracil/leucovorin plus bevacizumab (5 or 10 mg/kg) or placebo, every 2 weeks. Interestingly, the objective response rate was superior for the lower bevacizumab dose (40%), compared to the higher dose (24%), or placebo (17%). A second phase II trial in CRC then commenced dosing bevacizumab 10 mg/kg infusion every 2 weeks in combination with 5-fluorouracil/leucovorin/irinotecan (IFL). Overall the complete response rate, partial response rate, and stabilization rate were 5.4%, 38%, and 36% respectively, giving an overall disease control rate of 79% [10].
In phase III trials, in patients with mCRC, a median overall survival (OS) increase of 20.3 months was demonstrated with bevacizumab plus IFL, compared to 15.6 months with IFL alone [11]. In NSCLC, an OS of 12.3 months was observed with paclitaxel/carboplatin (PC) plus bevacizumab, compared to 10.3 months with PC alone [12]. In renal cancer, bevacizumab plus interferon alfa (IFN-α) improved median progression-free survival (PFS) by 89%: 10.2 months with bevacizumab plus IFN-α, compared to 5.4 months with IFN-α alone [13]. Median OS with bevacizumab plus IFN-α was 23 months, a non-significant increase compared to 21 months with IFN-α. In cervical cancer, OS increased to 16.8 months with bevacizumab plus chemotherapy, compared to 12.9 months with chemotherapy alone [14]. In platinum-resistant ovarian cancer, median PFS increased to 6.8 months with bevacizumab plus chemotherapy, compared to 3.4 months with chemotherapy alone [15]. In glioblastoma, bevacizumab showed an improvement in the objective response rate of 26% with a median duration response of 4.2 months compared to placebo; there are no data demonstrating an improvement in disease-related symptoms or increased survival [16]. A recently completed phase III trial in patients with malignant pleural mesothelioma, a rare cancer often diagnosed in people who have been exposed to asbestos, showed that adding bevacizumab to the standard treatment, pemetrexed and cisplatin, resulted in longer survival with acceptable toxicity [17].
5 Sorafenib (Nexavar®)
Sorafenib (5) is a tyrosine kinase inhibitor which was initially codeveloped by Bayer and Onyx as an inhibitor of c-RAF in the proliferative RAF/MEK/ERK MAP kinase pathway, but ultimately reached clinical success targeting VEGFR and platelet-derived growth factor receptor (PDGFRβ) [18]. It was the first small molecule approved by the FDA for the treatment of renal cell carcinoma in 2005, with a subsequent approval 2 years later for hepatocellular carcinoma (HCC), the most common form of liver cancer [1]. Clinical trials of sorafenib against melanoma, based primarily on its inhibition of the RAF kinase pathway, were unsuccessful [19].
The initial hit discovery took place in 1995 starting from the phenyl-urea thiophene ester 1 which was identified from a high-throughput screen against the c-RAF/MEK/ERK kinase cascade (see Fig. 3) [18]. The medicinal chemistry was optimization based around improving the c-RAF activity. This hit had moderate activity against c-RAF (IC50 17 μM) and a tenfold improvement was observed by 4-methyl substitution on the phenyl ring yielding 2. A library of bis-aryl urea analogs of the lead compound was then constructed to further explore the structure–activity relationship (SAR) of the series which identified the 3-amino-isoxazole 3 with c-RAF kinase inhibition IC50 of 1.1 μM. A fivefold increase in c-RAF activity was achieved by replacing its distal ring with a 4-pyridine 4, which also decreased lipophilicity, improved the aqueous solubility, and imparted significant inhibitory activity against human colon carcinoma HCT116 cell proliferation. Compound 4 possessed oral bioavailability and inhibited the growth of HCT116 xenografts in vivo, thereby providing proof of principle for this new kinase inhibitor class. Further SAR studies were then undertaken including evaluation of aromatic replacements of the isoxazole moiety, which identified the para-choro-meta-trifuroromethylphenyl ring as optimal for potency. Incorporation of a carboxamide functionality adjacent to the pyridyl ring further increased potency, with small groups such as methyl showing the best activity. These modifications led to the identification of sorafenib (5) with c-RAF kinase inhibition IC50 of 0.006 μM. Further profiling revealed that sofarenib potently inhibits b-RAF, as well as VEGFR-2 (IC50 = 0.09 μM), PDGFRβ (IC50 = 0.057 μM), RET kinase (IC50 = 0.006 μM), and a number of other kinases.


Key steps leading to the discovery of sorafenib (5)
Crystallography detailing the structure of various VEGFR-2 and inhibitor complexes has allowed for a detailed understanding of their binding modes and contributed greatly to drug design. Initial attempts to crystallize the catalytic kinase domain of VEGFR-2 containing the highly charged kinase-insert domain (KID) (see Fig. 1) failed to produce crystals after exhaustive screening. Crystallization was achieved using a construct in which the central 50 residues of the KID were deleted, leaving a loop of 20 residues to mimic the length of the analogous loop in the structure of FGFR-1 kinase. This truncated phosphorylated construct crystallized much more readily, producing a structure of the apo catalytic domain [20]. This construct was then used to obtain co-crystal structures of unphosphorylated kinase complexed with inhibitors. In the co-crystal structure of sorafenib bound to VEGFR-2, the nitrogen of the 4-pyridyl group of sorafenib accepts a hydrogen bond from the backbone NH of Cys919 in the hinge, whereas the methylamide NH donates a hydrogen bond to the carbonyl of Cys919, as shown in Fig. 4 [21]. It is interesting to note that the pyridyl moiety was introduced late in the lead optimization stage, meaning that the initial hit and early leads (1–3) did not possess the hinge binding motif. The inhibitor binds to the DFG-out or type II inactive conformation of the kinase, characterized by an almost 180° rotation of the conserved Asp-Phe-Gly (DFG) motif at the start of the activation loop in the ATP-binding cleft, relative to the active form, and a shift in the αC-helix. This creates a hydrophobic pocket (DFG-out pocket) that is occupied by the aryl-urea portion of sorafenib, with the lipophilic trifluoromethyl group of sorafenib occupying an additional small hydrophobic pocket. The urea group makes a pair of hydrogen bonding interactions, firstly with the backbone of Asp1046 from the DFG on the activation loop, and secondly a bidentate interaction with the acid side chain of Glu885 located in the middle of the αC helix, respectively.


Sorafenib (5) binding in the ATP pocket of VEGFR-2 kinase domain
In a phase I clinical trial of sorafenib in patients with advanced refractory solid tumors, the maximum-tolerated dose was 400 mg twice daily, with dose-limiting toxicities of diarrhea, fatigue, and grade 3 skin toxicity observed at higher doses [22]. Of the 45 patients assessed for efficacy, 1 hepatocellular carcinoma patient had a partial response, 25 patients had stable disease, with 8 lasting over 6 months and 5 for over 12 months.
Sorafenib was evaluated in a phase II trial patients with metastatic renal cell carcinoma (mRCC) [23]. The PFS was 24 versus 6 weeks in favor of sorafenib. The high rate of mRCC patients who were progression-free after 12 weeks of dosing led to its approval for this indication by the FDA. In a subsequent phase III study, the OS of patients receiving sorafenib was comparable with that of patients receiving placebo (17.8 versus 15.2 months); however, when crossover placebo survival data were censored, the difference became significant (17.8 versus 14.3 months, respectively) [24]. These results established the efficacy and safety of sorafenib in advanced mRCC.
In preclinical experiments, sorafenib had anti-proliferative activity in liver-cancer cell lines and it reduced tumor angiogenesis and increased tumor cell apoptosis in a mouse xenograft model of human hepatocellular carcinoma (HCC) [25]. It is known that the RAS/RAF/MEK/ERK pathway plays a role in HCC and that such tumors are highly vascularized and VEGF augments HCC development and metastasis. This provided a good rationale for investigating sorafenib for this indication. In a randomized phase III study the median OS increased from 7.9 months in the placebo group to 10.7 months in the sorafenib group [26]. On the basis of these findings, sorafenib was approved for the treatment of advanced HCC.
Sorafenib increased PFS for patients with radioactive iodine–refractory advanced thyroid cancer in a phase III trial, which supported the approval of sorafenib in this indication in 2013 [27]. Median PFS was 10.8 months in the sorafenib group, versus 5.8 months for placebo. PFS was improved with sorafenib in all clinical and genetic biomarker subgroups, irrespective of mutation status. RET kinase, BRAF V600E mutations, RAS mutations, and increased expression of VEGF and its receptors VEGFR have all been implicated in the pathogenesis and poor outcome of thyroid carcinoma; so it is likely the multi-kinase profile of sorafenib is contributing to its efficacy with this group.
6 Sunitinib (Sutent®)
The development of Sunitinib (10) began in 1994 at Sugen with a high-throughput screen of PDGFR kinase, which identified the indolin-2-ones 6 and 7 shown in Fig. 5, with moderate potency against PDGFR and additional potency against VEFGR-2 [28]. Cellular efficacy was observed in VEGF-induced human umbilical vein endothelial cell (v-HUVEC) signaling, compared to that induced by bFGF (b-HUVEC). Expansion of the SAR of this series led to the discovery of 8 (SU5416). SU5416 had no direct cytotoxic effects on tumor cell lines in vitro, but inhibited tumor vascular density and vascular leakage, suggestive of an anti-angiogenesis mechanism for its antitumor activity. In 1997, SU5416 became the first small molecule anti-angiogenic agent to enter the clinic, dosed by IV administration. However, despite evidence of clinical anti-angiogenesis activity, limitations in its pharmacokinetics and solubility profile restricted its further development. The addition of the propionic acid functionality attached to the dimethylpyrrole, as represented by 9, both served to increase the activity against PDGFR and improve the aqueous solubility and orally bioavailability of the series. The combination of both PDGFR and VEGFR activity for 9 translated to increased efficacy in mouse models of tumor xenografts compared to SU5416, and it was evaluated in the clinic as an oral drug in 1999. To further improve the developability profile, additional optimization at the dimethylpyrrole led to the identification of sunitinib (10). This contained the optimal profile in terms of potency, solubility, protein binding, pharmacokinetic properties, and antitumor efficacy.


Key steps leading to the discovery of sunitinib (10)
Compared with the binding of the type II inhibitor sorafenib deep into the pocket, sunitinib binds towards the front of the ATP site as shown in Fig. 6, leaving the interior unfilled [21]. The indole NH makes a hydrogen bond donation to the backbone carbonyl of Glu917 at the hinge of the kinase, whereas the carbonyl oxygen of the indole accepts a hydrogen bond from the NH backbone of Cys919. The dimethylamine function attached to the pyrrole ring is orientated out towards the solvent exposed region.


Sunitinib (10) binding in the ATP pocket of VEGFR-2 kinase domain
Sunitinib had good potency against VEGFR-1-3 and PDGFRβ (see Fig. 5) as well as the structurally related tyrosine kinases c-Kit, RET, and CSF1R [28]. It inhibited cellular proliferation of v-HUVECs (IC50 = 0.004 μM) and serum stimulated endothelial tube formation of human microvascular endothelial cells (IC50 = 0.055 μM). Sunitinib demonstrated marked tumor regression in mouse tumor xenograft models (e.g., HT29 colon carcinoma, A431 epidermoid carcinoma) with no direct cytotoxic effects observed against these tumor cell lines in vitro, supportive of the anti-angiogenic mechanism. Evaluation at different doses in these xenograft studies for tumor inhibition/regression, as well as inhibition of PDGFRβ and VEGFR-2 phosphorylation in tumor lysates, demonstrated that a minimal effective plasma concentration of 50 ng/mL was required to achieve full efficacy.
In the clinical setting, a phase I trial of patients with advanced solid tumors identified 50 mg/day (administered on a 4 weeks on and 2 weeks off schedule) as the recommended dose [29]. This achieved a median peak plasma concentration (Cmax) at steady state of 100–125 ng/mL (approximately 0.01 μM unbound), achieving adequate target plasma concentration to support further studies. In phase I studies assessing sunitinib as a single agent, from 117 patients there were a total of 16 confirmed objective partial responses, including 4 in metastatic renal cell carcinomas (mRCC), 4 in gastrointestinal stromal tumors (GIST), and 2 in tumors of neuroendocrine origin [30]. The GIST activity can be attributed to the inhibition of c-Kit by sunitinib, since GIST tumors frequently contain activating gene mutations in c-Kit. Sunitinib was shown to inhibit mutated variants of KIT (e.g., T670I, V654A) that are associated with imatinib resistance and tumor progression. Subsequent phase III clinical trials were carried with patients with mRCC, imatinib-refractory GIST, and neuroendocrine tumors. In mRCC patients, the PFS was significantly longer in the sunitinib group compared to interferon alpha therapy (11 versus 5 months) [31]. In patients with advanced GIST who failed on imatinib, the median time to tumor progression was 27.3 weeks with sunitinib, compared to 6.4 weeks for placebo [32]. In patients with pancreatic neuroendocrine tumors, PFS was increased to 11.4 months for sunitinib treatment, as compared with 5.5 months for placebo [33]. This led to the approval of sunitinib indicated for patients with advanced RCC and imatinib-resistant/intolerant GIST cancers in 2006, and subsequently neuroendocrine pancreatic tumors in 2011.
7 Pazopanib (Votrient®)
Pazopanib (17) was developed by GlaxoSmithKline and approved by the FDA for the treatment of renal cell carcinoma in 2009, with a subsequent approval 3 years later for soft tissue sarcoma [1]. The initial hit discovery took place in the late 1990s starting from two distinct screening hits: the moderately potent dianilino-2,4-pyrimidine 11 (VEGFR-2 IC50 = 0.4 μM), and the more potent 4-anilino-6,7-dimethoxyquinazoline 12 (VEGFR-2 IC50 = 0.006 μM), shown in Fig. 7 [34]. Combination of the key structural features of these generated N-(3-bromoanilino)-N′-(4-methyl-3-hydroxyanilino)-2,4-pyrimidine (13) (VEGFR-2 IC50 = 0.006 μM), with a cellular potency inhibiting v-HUVEC cells of IC50 = 0.54 μM. The pharmacokinetic profile of 13 and similar analogs was poor, presumably due to rapid phase II glucuronidation or sulfation reactions of the phenol functionality. Replacing the phenol with a 3-methylindazole heterocycle yielded the indazolylpyrimidine 14, which possessed both good potency against VEGFR-2 (IC50 = 0.006 μM) and v-HUVECs (IC50 = 0.18 μM), combined with significantly improved pharmacokinetics, with a clearance of 16 mL/min/kg and an oral bioavailability of 85% at a dose of 10 mg/kg in the rat.


Key steps leading to the discovery of pazopanib (17)
Inhibitor 14 was assessed in an in vivo efficacy study using HT29 human colon tumor xenografts in nude mice, and doses of 30 or 100 mg/kg over 23 days resulted in a 53 and 91% reduction in tumor size, respectively [34]. The N-methylated pyrimidine 15 showed an improved pharmacokinetic profile in the rat with lower clearance (10 mL/min/kg) and higher oral bioavailability (65%). At this stage, pyrimidines containing the 3-methylindazole heterocycle and methylated at the C-4 amino nitrogen possessed both good in vitro and cross-species pharmacokinetic profiles. However, significant inhibition (IC50 < 10 μM) was observed against a number of cytochrome P450 (CYP) isozymes, in particular 2C9. This presumably results from binding of the indazole nitrogens to the heme iron of the CYP enzyme. This activity was diminished by methylation of the indazole 2-nitrogen leading to 2,3-dimethylindazole becoming the preferred heterocycle, as represented by 16. A final optimization of the aniline ring led to the identification of pazopanib (17) with the optimal combination of in vitro potency and pharmacokinetic developability profiles.
Pazopanib is an ATP-competitive inhibitor of VEGFR-2 with a Ki of 24 nM in an in vitro assay. In cellular assays, pazopanib inhibited proliferation of v-HUVECs with an IC50 of 0.021 nM, with a 35-fold selectivity over b-HUVEC proliferation [34]. It also inhibited VEGF-induced phosphorylation of VEGFR-2 in HUVEC cells with an IC50 of ~8 nM. A greater than 1400-fold selectivity was observed against a variety of tumor cells and greater than 48-fold against fibroblasts as compared with v-HUVEC proliferation. Pazopanib possessed good pharmacokinetics in rat, dog, and monkey with low clearance, low volume of distribution, and good oral bioavailability.
The pazopanib co-crystal structure in VEGFR-2 shows that it binds in the ATP pocket (Fig. 8) [21]. The pyrimidine N-1 and the C-2 anilino N-H make hydrogen acceptor and donor interactions with the peptide backbone of Cys919 at the hinge. The 2,3-dimethylindazole head group pushes into the back pocket of the ATP site, whilst the NH of the sulfonamide makes a hydrogen bond interaction with Lys920.


Pazopanib (17) binding in the ATP pocket of VEGFR-2 kinase domain
As a measure of its ability to inhibit angiogenesis in vivo, pazopanib was examined in the mouse matrigel plug assay [35]. In this assay a gel plug of extracellular matrix containing b-FGF is implanted subcutaneously to stimulate vascularization inside the plug. Following once-daily oral administration of pazopanib for 5 days, the plug was removed and angiogenesis, as determination by the hemoglobin content, was inhibited in a dose-dependent manner, at doses from 10 to 100 mg/kg. Pazopanib was also examined in a second animal model of angiogenesis, the mouse corneal micropocket assay, in which ocular angiogenesis is induced by implantation of slow release pellets of VEGF into the mouse cornea. Treatment of mice with 100 mg/kg of pazopanib twice daily for 5 days resulted in significant inhibition in the degree of vascularization. The anti-angiogenic activity of pazopanib was demonstrated in mice bearing established human xenografts using HT29 (colon carcinoma), A375P (melanoma), and HN5 (head and neck carcinoma) tumors, showing a clear dose response following daily doses from 10 to 100 mg/kg over 3 weeks.
To establish a pharmocokinetic-pharmodynamic (PK-PD) correlation, the effect of pazopanib on VEGF-induced VEGFR-2 phosphorylation in vivo was evaluated in mouse lungs, which were chosen due to their high endothelial cell content [35]. A single oral dose of 30 mg/kg pazopanib inhibited phosphorylation for more than 8 h, corresponding to >40 μM plasma concentration. At 16 and 24 h, the plasma concentration dropped below 40 μM and the inhibition of VEGFR-2 phosphorylation was minimal, indicating that a ≥40 μM steady state concentration of pazopanib is required for optimal in vivo activity.
A phase I study evaluating safety and tolerability led to a recommended dose of 800 mg daily [36] with the most common adverse events being nausea, diarrhea, anorexia, hypertension, fatigue, hair depigmentation, and vomiting. A phase II study evaluating patients with advanced renal cell carcinoma (RCC) showed a 35% response to treatment, with stable disease achieved in 45% of patients [37]. Responses were durable with median duration of 68 weeks and an estimated median PFS of 11.9 months. In a phase III trial patients with advanced and/or metastatic RCC, PFS was significantly prolonged with pazopanib compared with placebo in the overall study population (9.2 versus 4.2 months) [38]. In the treatment-naive subpopulation a median PFS of 11.1 versus 2.8 months was observed, compared to the cytokine-pretreated subpopulation median PFS of 7.4 versus 4.2 months. The objective response rate was 30% with pazopanib compared with 3% for placebo, and the median duration of response was longer than 1 year. This led to the approval of pazopanib indicated for patients with RCC in 2009.
A phase II study was conducted in patients with advanced and/or metastatic soft tissue sarcoma (STS) who had relapsed following standard therapies [39]. The progression free rate for patients after 12 weeks was 44% with leiomyosarcoma, 49% with synovial sarcomas, and 39% in other STS types, but no activity observed in adipocytic STS. In a subsequent phase III study, patients were randomly assigned to receive pazopanib or placebo, and the median PFS was 4.6 months for pazopanib compared with 1.6 months for placebo [40]. Overall survival was 12.5 months with pazopanib, versus 10.7 months with placebo. This led to the additional approval of pazopanib for patients with soft tissue sarcomas in 2012.
8 Second Generation Marketed Anti-Angiogenesis Inhibitors in Oncology
With the approval of the VEGF monoclonal antibody bevacizumab in 2004, followed by the three small molecule kinase inhibitors, sorafenib, sunitinib, and pazopanib, and their adoption in clinical practice, the clinical path for subsequent VEGFR inhibitors became more challenging. The next generation of VEGFR inhibitors (Table 2) would need to demonstrate either superior efficacy over these in their respective indications or show efficacy in indications not covered by these approved drugs. For example, axitinib was approved in 2012 after showing extended PFS when compared to sorafenib in patients with advanced kidney cancer, but only as a second line treatment [1]. Regorafenib was approved in the same year for mCRC cancer, the first small molecule VEFGR-2 inhibitor for this indication, and the following year for advanced gastrointestinal stromal tumors, but only for patients resistant to imatinib and sunitinib [1]. Nintedanib is only FDA approved for a non-oncology indication (idiopathic pulmonary fibrosis), but has been approved in Europe in 2015 for combination therapy in NSCLC (retrieved from http://www.ema.europa.eu/ema). Lenvatinib was approved in 2015 for the treatment of radioactive iodine refractory differentiated thyroid cancers, 2 years after the approval for sorafenib for the same indication [1]. Vandetanib and cabozantinib have both been approved for late-stage (metastatic) medullary thyroid cancer [1]. Although both are multi-kinase inhibitors, including VEGFR-2, their mechanism of action is thought to be primarily inhibition of RET kinase, since both hereditary germline and somatic mutations in the RET proto-oncogene are known to cause medullary thyroid carcinomas. From this perspective their development falls out of the scope of this chapter. Ramucirumab is a VEGFR-2 antibody recently approved as second line treatment for advanced gastric cancer, metastatic NSCLC, and metastatic renal cell carcinoma [1].
9 Axitinib (Inlyta®)
Axitinib (23) was developed at Pfizer using a structure-based drug design approach, relying on solved co-crystal structures to guide ligand design, combined with a focus on optimizing binding efficiencies [41]. Screening identified the pyrazole 18 with moderate potency against VEFGR-2, which was selected over other hits for optimization based on superior ligand efficiency (Fig. 9). The initial goals were to improve potency, increase permeability, and remove the metabolic labile aryl hydroxy and methoxy groups. Two strategies were employed, first to truncate the molecule to determine the key features that exhibited high efficiency, and second to introduce conformational constraints to lock the molecule into the preferred bound conformation. Together this led to the identification of indazole 19 which became the core pharmacophore. Adding back the styrene functionality led to the full-length indazole possessing excellent potency and ligand efficiency, but poor kinase selectivity and low oral exposure in mouse. The co-crystal structures of indazole 20 revealed that it binds to the active DFG-in conformation, providing a rationale for the broad kinase activity observed. By contrast, other hits which bound the DFG-out conformation demonstrated a superior kinase selectivity profile, leading the team to design DFG-out features into this indazole series. Attachment of a phenyl thiol functionality at the 6-position of the indazole ring, as represented by 21, achieved the targeting of the DFG-out conformation, whilst also removing the metabolic aryl hydroxy and methoxy liabilities. Adding back the styrene functionality gave 22 with significant improved potency. Although 22 is less potent than 20, it possesses similar overall ligand efficiency and a superior kinase selectivity. Addition of a N-methylcarboxamide at the ortho position of the phenyl thiol, adding critical hydrogen-bond interactions in the DFG-out conformation, and replacement of the terminal phenyl with ortho-pyridine, led to axitinib (23) with pico-molar potency and lowered lipophilicity.


Key steps leading to the discovery of axitinib (23)
The solved structure of axitinib bound to VEGFR-2 shown in Fig. 10 provides insight into how the indazole series induced the DFG-out conformation with excellent efficiency [21]. The indazole core makes two hydrogen bond interactions to the hinge, the NH donating to the backbone carbonyl of Glu917 and the nitrogen accepting from the NH backbone of Cys919. The styryl group fills the narrow tunnel as it extends out toward the solvent front. Compared with the other inhibitors, the axitinib phenyl thiol head group is positioned slightly higher and deeper into the back pocket. The carboxamide forms one direct H-bond to the NH backbone of Asp1046 and a second direct H-bond to the carboxylate side chain of Glu885. The head group substantially complements the full length of the channel, contributing to high affinity with both polar charge stabilization and hydrophobic interactions.


Axitinib (23) binding in the ATP pocket of VEGFR-2 kinase domain
Axitinib demonstrated antitumor activity in a range of human tumor models in mice, including colon, lung, breast, pancreas, kidney, brain, melanoma, and hematopoietic malignancy. In the first study of axitinib in patients with advanced cancer, axitinib dosed 5 mg orally twice daily, showed evidence of clinical activity, with sustained tumor response seen in patients with RCC as well as adenoid cystic cancer [42]. Hypertension, a common side effect in VEGFR-2 targeted therapies, was the most frequently reported adverse effect. A phase II study showed activity in patients with metastatic renal cell carcinoma (mRCC) refractory to sorafenib (including subgroups refractory to both sunitinib and sorafenib, or to cytokines and sorafenib) [43]. A phase III study of 723 patients with advanced RCC randomized to receive either axitinib or sorafenib as second-line treatment showed a PFS of 8.3 months for axitinib compared to 5.7 months for sorafenib [44]. Overall survival, a secondary endpoint for the study, did not differ between the two groups. Based on this axitinib was approved for the second-line treatment of patients with advanced RCC in 2012.
A phase II clinical trial showed good response in combination chemotherapy with gemcitabine for advanced pancreatic cancer [45]. However a phase III trial comparing axitinib plus gemcitabine with gemcitabine alone for this patient population found no evidence of improvement in the primary endpoint of survival with the combination [46].
Additional phase II studies in patients with refractory thyroid cancer, NSCLC, and metastatic melanoma have been carried out with evidence of efficacy. At the time of writing, trials were recruiting for patients with melanoma and neurofibromatosis type 2, whilst trials for liver cancer, soft tissue sarcomas, and nasopharyngeal carcinomas were active.
10 Regorafenib (Stivarga®)
Regorafenib (24) is a follow-up to sorafenib codeveloped by Bayer and Onyx [47]. It is structurally very similar to sorafenib, differing by only addition of a fluorine atom ortho to the urea at the central phenyl group (Fig. 11). Biochemically this leads to a ~20-fold increase in potency for regorafenib against VEFGR-2 compared to sorafenib.


Comparison of regorafenib (24) with sorafenib (5)
Regorafenib demonstrated inhibition of tumor cell proliferation in various cell lines, with most notable activity in Kit and RET-mutated cell lines, including GIST 882 and thyroid TT cancer cells, with IC50 values of 45 and 34 nM, respectively [47]. Dose-dependent antitumor activity has been documented in colorectal (COLO 205), breast (MDA-MB-231), and renal cell (786-0) cancer xenograft models in mice.
Regorafenib was assessed as a single-agent in a phase I study in patients with a variety of tumors who had received multiple prior treatments [48]. Grade 3–4 adverse events did occur with hand-foot skin reaction, hypertension, diarrhea, rash, and fatigue being most frequently noted and the final recommended dose was 160 mg daily for 21 days in a 28-day cycle.
The approval for regorafenib in metastatic colorectal cancer was based on the results of a phase III trial of patients with who had previously received standard chemotherapies, including fluoropyrimidine, oxaliplatin, irinotecan, bevacizumab, and, if they were KRAS exon 2 wild-type, the anti-EGFR therapies panitumumab or cetuximab [49]. Regorafenib improved prolongation in OS in patients from 5 to 6.4 months compared to the placebo group. The trial also demonstrated an improvement in PFS from 1.7 to 2.0 months.
Regorafenib was subsequently approved for patients with advanced GIST that cannot be surgically removed and no longer respond to imatinib and sunitinib. The approval was based on the results of a phase III trial of patients with metastatic or unresectable GIST who experienced disease progression on, or were intolerant to, imatinib and sunitinib [50]. The primary endpoint was PFS, which increased from 0.9 to 4.8 months for patients who received regorafenib compared to placebo. Overall survival was not significantly different between the two groups, although 85% of patients in the placebo group ultimately crossed over to the regorafenib group.
A phase II study evaluating regorafenib in patients with advanced hepatocellular carcinoma who had progressed on sorafenib therapy demonstrated efficacy and a manageable safety profile [51]. A follow-up phase III trial was ongoing at the time of writing, and its results will determine if regorafenib can be approved for this type of liver cancer.
11 Nintedanib (Ofev®/Vargatef®)
Nintedanib (27) was developed starting from cross-screening of compounds prepared for a CDK4 program at Boehriger Ingelheim, which identified the 6-amido-substituted indolinone 25 (Fig. 12) as a sub-micromolar inhibitor of VEGFR-2 [52]. Interestingly, 25 itself had no CDK4 activity and showed a favorable kinase selectivity profile. Replacing the central aryl group with smaller alkyl substituents was detrimental to the chemical stability, probably because the phenyl group adopts a strain-free conformation perpendicular to the rest of the molecule (as shown Fig. 13 for the nintedanib co-crystal structure in VEGFR-2). The central aryl group also improved aqueous solubility, likely because it disrupts the overall flat shape and reduces aromatic-aromatic stacking in the crystalline state. Substitution at the 6-position of the oxindole core had a significant impact on potency and the overall kinase selectivity profile. 6-Chloro-substitution was favorable, but led to poorer selectivity profiles. The 6-methyl ester substituted oxindoles, such as 26, gave good potency and kinase selectivity and surprisingly demonstrated acceptable oral exposures in rodents. At the other end of the molecule, the basic amine functionality substituted para to the anilino moiety gave a flat SAR profile since it is orientated towards the solvent front of the kinase pocket, but it was a useful handle to fine-tune cell selectivity and solubility. After evaluation in xenograft experiments, nintedanib (27) was chosen to progress into preclinical development in 2001.


Key steps leading to the discovery of nintedanib (27)


Nintedanib (27) binding in the ATP pocket of VEGFR-2 kinase domain
Nintedanib binds to the ATP-binding site in the hinge region with the oxindole forming two hydrogen bonds to the backbone nitrogen of Cys919 and the backbone carbonyl oxygen of Glu917, as shown in Fig. 13 [53]. This oxindole binding mode resembles closely that of sunitinib (see Fig. 6). The methyl piperidine is directed towards the solvent region with the 4-nitrogen atom forming a bidentate ionic interaction with the carboxylate oxygens of Glu850. The carbonyl of the methyl ester at the 6-position of the oxindole forms a hydrogen bond with Lys868.
Nintedanib gave significant plasma levels after oral administration in various species but is rapidly cleared from plasma by ester hydrolysis at later points in time [53]. Part of the in vivo efficacy may be attributed to a sustained duration of VEGFR-2 inhibition, lasting up to 32 h after compound exposure. The mechanistic reason for this prolonged inhibition is not yet understood. It is possible that such prolonged receptor blockade in combination with a fast clearance in vivo may lead to efficacy combined with a clean safety profile. Nintedanib demonstrated significant antitumor activity in xenograft models such as NSCLC (Calu-6 cells), human renal cell carcinoma (RCC) (Caki-1), colorectal (HT-29), ovarian (SKOV-3), and prostate carcinoma (PAC-120). In H460 cell NSCLC xenografts, nintedanib also demonstrated synergistic effects in combination with the cytotoxic drugs docetaxel or pemetrexed.
A phase I study evaluated nintedanib combined with docetaxel in patients with advanced NSCLC. The maximum tolerated dose of nintedanib was between 150 and 200 mg twice daily in combination with docetaxel [52]. The dose-limiting toxicity was grade 3 hepatic enzyme elevations in both alanine and aspartate aminotransferase, and drug-related adverse events included neutropenia, leukopenia, fatigue, alopecia, and decreased appetite. Among evaluable patients, 26% had a partial response and 47% had stable disease. In a phase III study as second-line therapy in patients with NSCLC, patients received nintedanib 200 mg twice daily plus docetaxel 75 mg/m2 daily, or docetaxel plus placebo [52]. Nintedanib, when added to docetaxel, extended OS from 10.3 to 12.6 months for patients with adenocarcinoma, with 25.7% of patients surviving for 2 years or more with nintedanib plus docetaxel, compared to 19.1% for patients on placebo plus docetaxel. Nintedanib demonstrated a manageable adverse event profile and did not significantly increase discontinuation rates compared to docetaxel alone. Based on this study the EU granted approval for nintedanib for use as second-line therapy in combination with docetaxel in patients with NSCLC of adenocarcinoma tumor histology in 2015.
At the time of writing, Nintedanib is being investigated in a phase III study in patients with ovarian cancer and in phase II studies for mesothelioma and kidney cancers. It has recently completed phase III studies in advanced NSCLC and phase II trials in refractory colorectal and liver cancers.
12 Lenvatinib (Lenvima®)
Developed by Eisai, lenvatinib (28) potently inhibits VEGFR-1,2,3, FGFR-1, and RET tyrosine kinases and has recently been approved for patients with radioiodine-refractory differentiated thyroid cancers [54]. The efficacy against thyroid cancers is likely based on a combination of both VEGFR anti-angiogenic and RET antitumorigenic pathway inhibition. Lenvatinib inhibits RET kinase and VEGFR-2 kinase with Ki values of 1.5 and 0.74 nM, respectively (Fig. 14). Lenvatinib inhibited proliferation and phosphorylation of RET as well that of downstream ERK1/2 in medullary thyroid carcinoma cell lines, which expressed RET kinase in a dose-dependent manner. Significant antitumor activity was observed in mouse xenografts from these cell lines at doses of 10–100 mg/kg without affecting microvessel density, suggesting that activity was caused primarily by RET kinase inhibition. However, lenvatinib did also significantly inhibit in vivo growth of SW579 tumors in nude mice, a cell line which does not express RET kinase, accompanied by decreased microvessel density within the tumors, indicative of anti-angiogenic activity.


Kinase profile of lenvatinib (28)
A co-crystal structure revealed that the quinoline nitrogen of lenvatinib accepts a hydrogen bond from the hinge Cys919 as shown in Fig. 15 [55]. As observed with sorafenib, the urea makes two hydrogen bonding interactions, first with the backbone of Asp1046 from the DFG on the activation loop and second a bidentate interaction with the acid side chain of Glu885 located in the middle of the αC-helix. Interestingly, lenvatinib, in contrast to sorafenib, binds to the active DFG-in conformation. This is likely due to the much smaller cyclopropane urea substituent of lenvatinib located in the back of the pocket, compared to the 4-chloro-3-(trifluoromethyl) phenyl group of sorafenib. The polar carboxamide substituent at the 6-position of the quinoline ring is located towards the solvent front and forms a number of water mediated hydrogen bond interactions to Asn923.


Lenvatinib (28) binding in the ATP pocket of VEGFR-2 kinase domain
A phase I study of lenvatinib in Japanese patients with advanced solid tumors established an optimal daily dose of 24 mg [56]. In a phase II trial of lenvatinib in advanced radioiodine-refractory differentiated thyroid cancer (RR-DTC), patients who received lenvatinib 24 mg daily in 28-day cycles until disease progression had an objective response rate (ORR) of 50%, with only partial responses reported. The median time to response was 3.6 months, the median response duration was 12.7 months, and the median PFS was 12.6 months. The ORR for patients who had received previous VEGF therapy was 59%. Adverse events, regardless of their relation to treatment, occurred in 72% of patients and most frequently included weight loss, hypertension, proteinuria, and diarrhea.
In a randomized phase III study involving patients with RR-DTC given the same dose, lenvatinib treated subjects lived a median of 18.3 months without their disease progressing, compared to 3.6 months for subjects who received placebo [57]. Additionally, 65% of subjects treated with lenvatinib saw a reduction in tumor size, compared to 2% of subjects who received a placebo. The median OS was not reached in either group. Treatment-related adverse effects of all grades occurred in more than 40% of patients in the lenvatinib group, including hypertension, diarrhea, fatigue, decreased appetite and weight, and nausea. In the lenvatinib group, 6 of 20 deaths that occurred during the treatment period were considered to be drug-related. Based on this, lenvatinib was approved for patients with RR-DTC in 2015.
At the time of writing, Lenvatinib was also under phase III clinical investigation for hepatocellular carcinoma and phase II studies for NSCLC, biliary tract, and RCC cancers. Two phase II studies for melanoma and endometrial cancers have completed.
13 Third Generation Anti-Angiogenesis Inhibitors in Oncology Under Development
The third generation of VEGFR inhibitors under clinical development faces a prohibitively crowded field with multiple anti-angiogenesis drugs now approved and indicated for a wide range of tumor types. The clinical trials for these investigational drugs aimed at demonstrating superior efficacy over the approved VEGFR drugs have largely failed. Their focus would now appear to be on exploring new indications for VEGFR inhibitors, such as ovarian, gastric, and mesothelioma cancers, as well as NSCLC cancers driven by specific gene amplifications (Table 3).
14 Cediranib
Cediranib (29) is a potent inhibitor against VEGFR-1-3, c-Kit, and PDGFR kinases which is currently under investigation by AstraZeneca for ovarian cancer (Fig. 16) [58].


Kinase profile of cediranib (29)
Cediranib had been investigated by AstraZeneca in a number of advanced clinical trials, but the company had abandoned its development in 2011 after these trials were not successful [59]. For example, in a phase III trial for patients with mCRC cancer who had progressed following first-line therapy, addition of cediranib (20 or 30 mg daily) or bevacizumab to the combination chemotherapy oxaliplatin/leucovorin/5-fluorouracil showed no differences in PFS [60]. A demonstration of improvement over bevacizumab was required to move forward with registration. In a phase III trial comparing cediranib with carboplatin/paclitaxel chemotherapy in NSCLC patients, cediranib (20 mg daily) did increase the response rates (52% versus 34%), albeit with an increase in toxicity (grade 3 hypertension, diarrhea, and anorexia), but showed no increase in either PFS or OS [60]. In a phase II trial with first line treatment-naive renal cell carcinoma patients, cediranib demonstrated comparable anti-tumor activity, but no benefit over the approved VEGFR inhibitors sunitinib and pazopanib. In a phase III trial comparing cediranib with the alkylating agent lomustine for patients with recurrent glioblastoma, there was no significant difference in PFS or OS for either cediranib alone (30 mg daily) or cediranib/lomustine (20 mg daily) compared with lomustine alone [60].
However, recent investigator-initiated studies in gynecological cancers have revived interest in cediranib. In a trial sponsored by Cancer Research UK, cediranib combined with platinum-based chemotherapy improved PFS and OS in patients with relapsed ovarian cancer [59]. It was reported that cediranib (20 mg daily) had extended PFS by 3.2 months and OS by 2.7 months in combination with chemotherapy and in maintenance treatment, compared with chemotherapy alone. This is the first VEGFR inhibitor to demonstrate an OS benefit in recurrent ovarian cancer.
Investigators at the National Cancer Institute reported phase II results showing that cediranib combined with olaparib nearly doubled PFS in women with platinum-sensitive recurrent ovarian cancer or ovarian cancer associated with BRCA mutations [59]. Women taking the drug combination had PFS lasting 17.7 months, compared with 9 months among those given olaparib alone. However, these findings were tempered by grade 3 and 4 toxicities being higher with the drug combination, with reports of fatigue, diarrhea, and hypertension, in addition to more serious side effects, including myelodysplastic syndrome, weight loss, and one case of vaginal fistula.
AstraZeneca is now reportedly looking to move forward for regulatory approval for cediranib specifically in ovarian cancer [59]. Patients whose tumors are sensitive to platinum-based chemotherapy may offer an opportunity for cediranib, especially since bevacizumab’s approval is limited to platinum-resistant patients. At the time of writing, additional investigator-initiated trials were also underway in sarcoma and mesothelioma (Table 3).
15 Tivozanib
Tivozanib (30) is a very potent VEGFR inhibitor that is being codeveloped by Kirin Brewery and AVEO Pharmaceuticals with sub-nanomolar IC50 values against all three isoforms, as shown in Fig. 17 [61]. The drug has a very long half-life of more than 4 days (112 h) in humans. The exceptional potency and long half-life led to low doses of 1, 1.5, and 2 mg/day being explored for clinical use. Based on these studies, a maximum tolerated dose of 1.5 mg/day, 4 weeks on and 2 weeks off, was determined. At this dose, the adverse events observed were similar to other oral VEGFR inhibitors including hypertension, dysphonia, diarrhea, and abnormal liver functions.


Kinase profile of tivozanib (30)
A phase III trial compared tivozanib with sorafenib in patients with metastatic renal cell carcinoma (mRCC) who were either untreated or had received cytokines [62]. The study met its primary end point of improving PFS for tivozanib compared to sorafenib (11.9 versus 9.1 months), but showed a lowered OS (28.8 versus 29.3 months). Crossover of patients from sorafenib to tivozanib may have confounded the survival comparison. Because of the survival findings, the FDA rejected approval in May 2013 and requested additional clinical studies, casting uncertainty over its future development for mRCC.
A phase II trial in patients with stage IV mCRC cancer compared addition of tivozanib or bevacizumab to the combination chemotherapy oxaliplatin/leucovorin/5-fluorouracil [62]. An interim analysis showed comparable efficacy, but without the necessary demonstration of superiority over bevacizumab the trial was discontinued. However, one of the potential biomarkers explored in this study was neuropilin-1 (NRP-1), a signaling protein known to bind to VEGF-A in serum. NRP-1 was found to be a potential prognostic marker for angiogenesis inhibitor activity and could be predictive of tivozanib activity relative to bevacizumab [63]. One current proposal for further development of tivozanib is a subsequent trial targeting mCRC patients with low baseline expression of NRP-1. At the time of writing, an additional investigator-initiated trial was also currently underway in liver cancers (Table 3).
16 Dovitinib
Developed by Novartis, dovitinib (31) is a potent inhibitor against the VEGFR and FGFR isoforms, plus PDGFR (Fig. 18) [64]. The potent activity against FGFR in combination with VEGFR was the main rationale to evaluate dovitinib in patients who had relapsed with one of the more VEGFR selective first-generation VEGFR inhibitors.


Kinase profile of dovitinib (31)
In phase I studies, dovitinib showed accumulation at doses over 400 mg/day; thus a well-tolerated intermittent dosing schedule of 500 mg on a 5 days on, 2 days off schedule was adopted [64]. Antitumor activity was observed in pretreated patients with metastatic RCC. Two patients (13%) achieved a partial response, and the PFS and OS were 8.1 and 13.3 months, respectively. Pharmacodynamic analysis of plasma biomarkers and tumor biopsies showed both VEGFR inhibition (via increased PLGF levels and decreased sVEGFR-2 levels) and FGFR inhibition (via induction of FGF23, a biomarker of FGFR-1 inhibition), thus demonstrating clinical inhibition of both pathways. However, a phase III trial comparing dovitinib with sorafenib in patients with metastatic RCC, who had received one prior VEGF/VEGFR-targeted agent and one prior mTOR inhibitor, did not demonstrate superior efficacy [64]. Similar PFS (3.7 and 3.6 months) and OS (11.1 and 11.0 months) values were found for dovitinib and sorafenib, respectively. A phase II trial evaluated the efficacy of dovitinib in patients with metastatic GIST, after the failure of at least imatinib and sunitinib, showed only modest antitumor activity, with one patient (3%) showing a partial response. Dovitinib was being actively studied at the time of writing in patients with gastric cancer and relapsed glioblastoma, as shown in Table 3. More specific patient selection for tumors where FGFR amplification is observed may be the key to observe superior efficacy compared to the current standard of care treatments with this drug.
17 Apatinib
Apatinib (32) is a VEGFR-2 inhibitor being codeveloped by Jiangsu Hengrui Medicine (China), LSK BioPartners (USA), and Bukwang Pharmaceutical Company (Korea) (see Fig. 19) [65]. A phase I study in patients with advanced solid malignancies showed encouraging antitumor activity, with several patients achieving complete or partial responses, and a manageable toxicity profile leading to a maximum tolerated dose of 850 mg daily. A phase II⁄III study with apatinib (850 mg daily or 425 mg twice daily) was evaluated in patients with metastatic gastric cancer who had failed two lines of chemotherapy [65]. Progression-free survival was increased from 1.4 months for placebo to 3.67 and 3.2 months for single and twice daily dosed apatinib, respectively. The OS times were increased from 2.5 months for placebo to 4.83 and 4.27 months for single and twice daily dosed apatinib, respectively. Apatinib was approved by the Chinese FDA in December 2014 for patients with late-stage gastric cancer.


Kinase profile of apatinib (32)
At the time of writing, Apatinib was being studied in patients with esophageal, gastric, and liver cancers. Additional trials are also underway with advanced NSCLC patients with either RET fusion positive or wild-type EGFR tumors (Table 3).
18 Inhibitors No Longer Under Development
Despite the success of the field, a number of VEGFR-2 inhibitors have progressed to late stage clinical trials and have not met their primary endpoints and appear to be discontinued. Motesanib was being developed by Amgen and Takeda. It was evaluated in a phase III trial in combination with paclitaxel and carboplatin chemotherapy for NSCLC, but demonstrated no benefit on OS [66]. No further development of motesanib appears to have taken place.
Brivanib was being developed by Bristol-Myer Squibb (BMS), but recently failed in three randomized controlled phase III trials: a first-line trial in hepatocellular carcinoma (HCC) against sorafenib [67]; a second-line trial in HCC patients who were intolerant or failed with sorafenib [68]; and a combination trial (brivanib plus cetuximab vs cetuximab alone) in patients with metastatic, chemotherapy-refractory, wild-type KRAS colorectal carcinoma [69]. No further development of brivanib has since been reported by BMS.
Linifanib was developed by Abbott and was studied in patients with liver cancer in comparison to sorafenib. Linifanib and sorafenib had similar OS profiles with these patients, but linifanib did not meet predefined superiority and non-inferiority criteria and was discontinued [70].
Vatalanib, developed by Novartis, was studied in two separate phase III trials as combination with fluorouracil, leucovorin, and oxaliplatin for mCRC cancer, but had only a marginal effect on PFS and no detectable effect on OS [71]. No further development of vatalanib is currently reported by Novartis.
19 Conclusions
In a little over a decade following their introduction, the field of VEGF/VEGFR antiangiogenic agents appears to have reached maturity, with two antibodies and seven small molecule inhibitors of VEGFR are now approved for clinical use. The range of cancer indications they currently target includes liver, kidney, lung, ovarian, cervical, thyroid, pancreas, brain, soft tissue sarcoma, gastric, and GIST. New drugs being studied in the clinic appear to focus on lung, ovarian, gastric, and mesothelioma cancers. Novel combination therapies may provide additional opportunities to improve the clinical benefits of anti-angiogenesis drugs. Perhaps the most promising is the combination of antiangiogenic agents with the rapidly emerging field of immunomodulation. For example, multiple active clinical trials with the recently approved PD-1 inhibitors nivolumab (Opdivo®) and pembrolizumab (Keytruda®) with the approved drugs described in this chapter are listed in ClinicalTrials.gov. Identifying the most suitable agents, stand-alone or combination therapies, as well as the optimal dosage and schedules will continue to evolve as these drugs increasingly become standards of care in cancer therapy.
References
FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/drugsatfda/
Folkman J (2007) Nat Rev Drug Discov 6:273
Holmes K, Roberts OL, Thomas AM, Cross MJ (2007) Cell Signal 19:2003
Stuttfeld E, Ballmer-Hofer K (2009) IUBMB Life 61:915
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) Nat Rev Mol Cell Biol 7:359
Ferrara N (2011) Int J Dev Biol 55:383
Ferrara N, Hillan KJ, Gerber H-P, Novotny W (2004) Nat Rev Drug Discov 3:391
Gordon MS, Margolin K, Talpaz M, Sledge Jr GW, Holmgren E, Benjamin R, Stalter S, Shak S, Adelman D (2001) J Clin Oncol 19:843
Lu JF, Bruno R, Eppler S, Novotny W, Lum B, Gaudreault J (2008) Cancer Chemother Pharmacol 62:779
Keating GM (2014) Drugs 74:1891
Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, Hambleton J, Novotny WF, Kabbinavar F (2005) J Clin Oncol 23:3502
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2006) N Engl J Med 355:2542
Escudier B, Bellmunt J, Négrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V (2010) J Clin Oncol 28:2144
Tewari KS, Sill MW, Long 3rd HJ, Penson RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao MM, Michael HE, Monk BJ (2014) N Engl J Med 370:734
Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote I, Witteveen P, Bamias A, Pereira D, Wimberger P, Oaknin A, Mirza MR, Follana P, Bollag D, Ray-Coquard I (2014) J Clin Oncol 32:1302
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran Jr WJ, Mehta MP (2014) N Engl J Med 370:699
Zalcman G, Mazières J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, Molinier O, Corre R, Monnet I, Gounant V, Janicot H, Gervais R, Locher C, Milleron B, Tran Q, Lebitasy MP, Morin F, Creveuil C, Parienti J-J, Scherpereel A (2015) J Clin Oncol 33(Suppl); abstr 7500
Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S (2006) Nat Rev Drug Discov 5:835
Mangana J, Levesque MP, Karpova MB, Dummer R (2012) Exp Opin Invest Drugs 21:557
McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K (1999) Structure 7:319
McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS (2012) Proc Natl Acad Sci U S A 109:18281
Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, Schwartz B, Awada A, Voigtmann R, Scheulen ME, Seeber S (2005) J Clin Oncol 23:965
Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, Gore M, Desai AA, Patnaik A, Xiong HQ, Rowinsky E, Abbruzzese JL, Xia C, Simantov R, Schwartz B, O’Dwyer PJ (2006) J Clin Oncol 24:2505
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Anderson S, Hofilena G, Shan M, Pena C, Lathia C, Bukowski RM (2009) J Clin Oncol 27:3312
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C (2006) Cancer Res 66:11851
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) N Engl J Med 359:378
Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R, Shong YK, Sherman SI, Smit JW, Chung J, Kappeler C, Peña C, Molnár I, Schlumberger MJ (2014) Lancet 384:319
Sun CL, Christensen JG, McMahon G (2009) Discovery and development of sunitinib (SU11248): a multitarget tyrosine kinase inhibitor of tumor growth, survival, and angiogenesis. In: Li R, Stafford JA (eds) Kinase inhibitor drugs. Wiley, Hoboken, p. 1
Britten CD, Kabbinavar F, Hecht JR, Bello CL, Li J, Baum C, Slamon D (2008) Cancer Chemother Pharmacol 61:515
Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E (2006) J Clin Oncol 24:25
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA (2007) N Engl J Med 356:115
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Lancet 368:1329
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Hörsch D, Hammel P, Wiedenmann B, Van Cutsem E, Patyna S, Lu DR, Blanckmeister C, Chao R, Ruszniewski P (2011) N Engl J Med 364:501
Harris PA, Stafford JA (2009) Discovery of pazopanib, a pan vascular endothelial growth factor kinase inhibitor. In: Li R, Stafford JA (eds) Kinase inhibitor drugs. Wiley, Hoboken, p. 57
Kumar R, Knick VB, Rudolph SK, Johnson JH, Crosby RM, Crouthamel MC, Hopper TM, Miller CG, Harrington LE, Onori JA, Mullin RJ, Gilmer TM, Truesdale AT, Epperly AH, Boloor A, Stafford JA, Luttrell DK, Cheung M (2007) Mol Cancer Ther 6:2012
Hurwitz H, Dowlati A, Savage S, Fernando N, Lasalvia S, Whitehead B, Suttle B, Collins D, Ho P, Pandite L (2005) J Clin Oncol 23:3012
Hutson TE, Davis ID, Machiels JH, de Souza PL, Baker K, Bordogna W, Westlund R, Crofts T, Pandite L, Figlin RA (2008) J Clin Oncol 26:5046
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarbá JJ, Chen M, McCann L, Pandite L, Roychowdhury DF, Hawkins RE (2010) J Clin Oncol 28:1061
Sleijfer S, Papai Z, Le Cesne A, Scurr M, Ray-Coquard I, Collin F, Pandite L, Marreaud S, De Brauwer A, Blay J (2007) J Clin Oncol 25:10031
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schöffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, Araki N, Ouali M, Marreaud S, Hodge R, Dewji MR, Coens C, Demetri GD, Fletcher CD, Dei Tos AP, Hohenberger P (2012) Lancet 379:1879
Kania RS (2009) Structure-based design and characterization of axitinib. In: Li R, Stafford JA (eds) Kinase inhibitor drugs. Wiley, Hoboken, p. 167
Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G (2005) J Clin Oncol 23:5474
Rini BI, Wilding G, Hudes G, Stadler WM, Kim S, Tarazi J, Rosbrook B, Trask PC, Wood L, Dutcher JP (2009) J Clin Oncol 27:4462
Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, Oudard S, Gore ME, Tarazi J, Hariharan S, Chen C, Rosbrook B, Kim S, Rini BI (2013) Lancet Oncol 14:552
Spano J, Chodkiewicz C, Maurel J, Wong RP, Wasan HS, Pithavala YK, Bycott PW, Liau K, Kim S, Rixe O (2007) J Clin Oncol 25:4551
Kindler HL, Ioka T, Richel DJ, Bennouna J, Létourneau R, Okusaka T, Funakoshi A, Furuse J, Park YS, Ohkawa S, Springett GM, Wasan HS, Trask PC, Bycott P, Ricart AD, Kim S, Van Cutsem E (2011) Lancet Oncol 12:256
Davis SL, Eckhardt SG, Messersmith WA, Jimeno A (2013) Drugs Today 49:105
Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, Unger C, Krätzschmar J, Heinig R, Boix O, Christensen O (2012) Clin Cancer Res 18:2658
Grothey A, Sobrero AF, Siena S, Falcone A, Ychou M, Lenz H-J, Yoshino T, Cihon F, Wagner A, Van Cutsem E (2012) J Clin Oncol 30(Suppl 4); abstr LBA385
Demetri GD, Reichardt P, Kang Y-K, Blay J-Y, Joensuu H, Maki RG, Rutkowski P, Hohenberger P, Gelderblom H, Leahy MG, von Mehren M, Schoffski P, Blackstein ME, Le Cesne A, Badalamenti G, Xu J-M, Nishida T, Laurent D, Kuss I, Casali PG (2012) J Clin Oncol 30(Suppl); abstr LBA10008
Bruix J, Tak WY, Gasbarrini A, Santoro A, Colombo M, Lim HY, Mazzaferro V, Wiest R, Reig M, Wagner A, Bolondi L (2013) Eur J Cancer 49:3412
Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, Wollin SL, Kaiser R (2015) J Med Chem 58:1053
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ (2008) Cancer Res 68:4774
Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, Uenaka T, Asada M (2008) Int J Cancer 122:664
Okamoto K, Ikemori-Kawada M, Jestel A, von König K, Funahashi Y, Matsushima T, Tsuruoka A, Inoue A, Matsui J (2014) ACS Med Chem Lett 6:89
Nakamichi S, Nokihara H, Yamamoto N, Yamada Y, Honda K, Tamura Y, Wakui H, Sasaki T, Yusa W, Fujino K, Tamura T (2015) Cancer Chemother Pharmacol 76:1153
Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, Habra MA, Newbold K, Shah MH, Hoff AO, Gianoukakis AG, Kiyota N, Taylor MH, Kim SB, Krzyzanowska MK, Dutcus CE, de las Heras B, Zhu J, Sherman SI (2015) N Engl J Med 372:621
Sorbera LA, Serradell N, Rosa E, Bolós J, Bayés M (2007) Drugs Future 32:577
Schmidt C (2015) J Natl Cancer Inst 107:3
Sahade M, Caparelli F, Hoff PM (2012) Future Oncol 8:775
Haberkorn BC, Eskens FA (2013) Future Oncol 9:13
Jamil MO, Hathaway A, Mehta A (2015) Curr Oncol Rep 17:24
Benson A, Krivoshik A, Van Sant C, Needle M (2015) Ann Oncol 26(Suppl 4):108
Porta C, Giglione P, Liguigli W, Paglino C (2015) Future Oncol 11:39
Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y, Li J, Lou L (2011) Cancer Sci 102:1374
Scagliotti GV, Vynnychenko I, Park K, Ichinose Y, Kubota K, Blackhall F, Pirker R, Galiulin R, Ciuleanu TE, Sydorenko O, Dediu M, Papai-Szekely Z, Banaclocha NM, McCoy S, Yao B, Hei YJ, Galimi F, Spigel DR (2012) J Clin Oncol 30:2829
Johnson PJ, Qin S, Park JW, Poon RT, Raoul JL, Philip PA, Hsu CH, Hu TH, Heo J, Xu J, Lu L, Chao Y, Boucher E, Han KH, Paik SW, Robles-Aviña J, Kudo M, Yan L, Sobhonslidsuk A, Komov D, Decaens T, Tak WY, Jeng LB, Liu D, Ezzeddine R, Walters I, Cheng AL (2013) J Clin Oncol 31:3517
Llovet JM, Decaens T, Raoul JL, Boucher E, Kudo M, Chang C, Kang YK, Assenat E, Lim HY, Boige V, Mathurin P, Fartoux L, Lin DY, Bruix J, Poon RT, Sherman M, Blanc JF, Finn RS, Tak WY, Chao Y, Ezzeddine R, Liu D, Walters I, Park JW (2013) J Clin Oncol 31:3509
Siu LL, Shapiro JD, Jonker DJ, Karapetis CS, Zalcberg JR, Simes J, Couture F, Moore MJ, Price TJ, Siddiqui J, Nott LM, Charpentier D, Liauw W, Sawyer MB, Jefford M, Magoski NM, Haydon A, Walters I, Ringash J, Tu D, O’Callaghan CJ (2013) J Clin Oncol 31:2477
Cainap C, Qin S, Huang WT, Chung IJ, Pan H, Cheng Y, Kudo M, Kang YK, Chen PJ, Toh HC, Gorbunova V, Eskens FA, Qian J, McKee MD, Ricker JL, Carlson DM, El-Nowiem S (2015) J Clin Oncol 33:172
Sobrero AF, Bruzzi P (2011) J Clin Oncol 29:1938
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Harris, P.A. (2017). Inhibitors of Vascular Endothelial Growth Factor Receptor. In: Waring, M.J. (eds) Cancer II. Topics in Medicinal Chemistry, vol 28. Springer, Cham. https://doi.org/10.1007/7355_2016_28
Download citation
DOI: https://doi.org/10.1007/7355_2016_28
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75924-1
Online ISBN: 978-3-319-75926-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)