
Abstract
d-tagatose is produced from d-galactose by the enzyme l-arabinose isomerase (L-AI) in a commercially viable bioprocess. An active and stable biocatalyst was obtained by modifying chitosan gel structure through reaction with TNBS, d-fructose or DMF, among others. This led to a significant improvement in L-AI immobilization via multipoint covalent attachment approach. Synthetized derivatives were compared with commercial supports such as Eupergit® C250L and glyoxal-agarose. The best chitosan derivative for L-AI immobilization was achieved by reacting 4 % (w/v) d-fructose with 3 % (w/v) chitosan at 50 °C for 4 h. When compared to the free enzyme, the glutaraldehyde-activated chitosan biocatalyst showed an apparent activity of 88.4 U g −1gel with a 211-fold stabilization factor while the glyoxal-agarose biocatalyst gave an apparent activity of 161.8 U g −1gel with an 85-fold stabilization factor. Hence, chitosan derivatives were comparable to commercial resins, thus becoming a viable low-cost strategy to obtain high active L-AI insolubilized derivatives.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Enzymes are biocatalysts with excellent perspectives for their application in industry due to their high specificity, activity and stereo- and region selectivity under very mild conditions [16, 47]. However, their implementation in industry has been hampered by their lack of long-term operational stability, complex down-stream processing, difficult recovery and re-use of the enzyme [13, 33, 51]. The well-known moderate to low stability of soluble enzymes can be overcome through enzyme immobilization [23, 39]. Among all possible immobilization strategies, carrier-bound methods are generally chosen due to the variety of supports with different chemical and mechanical properties that are available or can be properly produced [21, 53]. For an industrial use of enzymes, in particular, an approach that enhances the enzyme binding to support is recommended [10]. Hence, multipoint covalent attachment scheme becomes a viable alternative on the grounds that it produces irreversible bondings between enzyme and support, thus allowing highly active, reusable and long-term stable insoluble biocatalysts to be produced without releasing enzyme molecules to the reaction mixture [3, 18].
On the other hand, l-arabinose isomerase (L-AI; EC 5.3.1.4) is an intracellular multimeric enzyme that in vivo catalyzes the isomerization of l-arabinose to l-ribulose. However, it can also isomerize d-tagatose, a rare sugar with several promising nutraceutical properties, from d-galactose [28]. The scaling-up of the isomerization reaction has proved to be of great technological and nutritional interest with inherent industrial application [8, 42]. Hence, the production of highly active and stable insoluble enzyme biocatalysts is mandatory for their use in industrial processes [33]. For the current research, L-AI from Enterococcus faecium DBFIQ E36 strain, isolated from raw cow milk, was purified up to electrophoretical homogeneity and employed to obtain insoluble biocatalysts. Briefly, this enzyme presents a homo-tetrameric structure with a MW of 187 kDa determined by gel filtration, a pI of 3.8, and was also characterized as a Mn2+ ion-activated enzyme [37, 56].
Furthermore, the use of low-cost supports such as calcium carbonate, chitin and chitosan represents an economical advantage over commercial resins [11]. In that way, chitosan appears as an excellent biopolymer for support synthesis to be employed in enzyme immobilization because it is an economic and biodegradable product that can be prepared in several ways, such as hydrogels, membranes, particles, fibers, scales, among others [22, 45]. Chemically, chitosan free amine groups acquire positive charge in the presence of acid solutions, thereby bringing countless new properties to chitosan such as metal chelation and adsorption of enzymes and fats, among others [35]. Besides, the high number of free primary amine groups, directly related to the deacetylation degree, has allowed to chemically modify chitosan in a homogeneous way, based on the high nucleophilicity of amine groups in aqueous media [31].
Therefore, as L-AI is a metalloenzyme containing Mn2+, it is important to prevent cation adsorption by the support matrix while producing immobilized derivatives with improved stability and activity due to the multimeric nature of the enzyme. In addition, it has been proved that the binding of metal cations usually increases with pH, indicating that only the deprotonated amine groups can bind the ions [45]. In that way, several chemical modifications were performed on chitosan to obtain derivatives with reduced metal chelation properties. The improvement of chitosan properties as a support for metalloenzyme immobilization was achieved by modifying the polymer structure without changing the fundamental skeleton of the polymer [30].
According to several chemical modifications previously reported, different hydrogel structures were obtained by reacting chitosan, either pure or mixed, with other chemicals. First of all, chitosan primary amino groups undergo Schiff reaction with aldehydes or ketones to yield the corresponding aldimines and ketimines, which are converted to N-alkyl derivatives upon hydrogenation with sodium borohydride [50]. The use of this approach has allowed the transformation of the linear chitosan polymer into a branched-chain derivative through reductive alkylation using d-lactose [15, 26] and d-fructose [19, 61] as target carbohydrates.
On the other hand, blending chitosan with different polyelectrolytes leads to an intermolecular interaction between two polyelectrolytes that alter the physicochemical properties of the target biopolymer. Therefore, hybrid hydrogels are produced through complexion with polyvinyl alcohol [4, 41] and through formation of ionically cross-linked complexes with sodium tri(poly)phosphate [5, 60] and aluminum polychloride [48]. Superficial primary amines react with TNBS or DMF, modifying the hydrophobicity of the bead surface. In this case, chitosan hydrophobicity was modified through N-acylation with N, N-dimethylformamide [63] and with 2,4,6-trinitrobenzene sulfonic acid [40]. Furthermore, to change porosity and mechanical resistance of native and modified chitosan particles, a treatment with the dialdehyde glyoxal was performed [62].
Hence, modified gel particles were tested under different activation and immobilization conditions to produce multipoint covalent attachment derivatives. Finally, insoluble biocatalysts were assessed in their physicochemical properties and compared with soluble enzyme and L-AI immobilized derivatives on commercial resins.
Materials and methods
Materials
Native chitosan (85.2 % deacetylation degree) was purchased from Polymar (Fortaleza, Ce, Brazil). Sodium tripolyphosphate (TPP; Na5P3O10) and sodium metaperiodate (NaIO4) were purchased from Merck KGaA (Darmstadt, Germany). Fully hydrolyzed polyvinyl alcohol (PVA; 99 % hydrolyzed, with a molecular weight of 145,000 g mol−1) was acquired from Merck OHG division (Hohenbrunn, Germany). 2,4,6-trinitrobenzenesulfonic acid or pycrilsulfonic acid (TNBS), N,N-dimethylformamide (DMF), sodium borohydride (NaBH4), 25 % (v/v) glutaraldehyde solution (GA), 40 % (v/v) glyoxal solution, aluminum polychloride (PAC), d-fructose, d-lactose, d-galactose, ammonium sulfate and d-tagatose were directly purchased from Sigma Chemical Co. (St. Louis, MO, USA). Low and high viscosity sodium alginate were provided by Kelco under the commercial brand of Kelgin (Chicago, USA). Commercial agarose microparticles (wet particle diameter: 45-165 µm), under the commercial brand of Sepharose CL-6B, were purchased from GE Healthcare (Uppsala, Sweden). Eupergit® C250L was acquired from Degussa Specialty Polymers, Röhm GmbH and Co. KG (Darmstadt, Germany). Commercial supports of glyoxyl-activated agarose gel with either 4 or 6 % agarose concentration, and with various degrees of activation [very high (VH)/high (H)/low (L)], were purchased directly to Agarose Bead Technologies (Madrid, Spain). Commercial supports had the following characteristics: HDG-4BCL: 4 % cross-linked agarose with a high density of glyoxyl groups (40–60 µmol glyoxyl/ml gel); LDG-6BCL: 6 % cross-linked agarose with a low density of glyoxyl groups (15–25 µmol glyoxyl/ml gel); VHDG-6BCL: 6 % cross-linked agarose with a very high density of glyoxyl groups (80–100 µmol glyoxyl/ml gel). Salts, buffers and all other reagents were of analytical grade.
Methods
Enzyme production and purification
l-arabinose isomerase (EC 5.3.1.4) crude cell-free extract was obtained from Enterococcus faecium DBFIQ E36 strain isolated from raw cow milk, following the procedure described by Manzo et al. [38]. Pure preparations of the enzyme were achieved through clarification by membrane filtration, ammonium sulfate precipitation (85 % saturation), followed by l-arabitol-Sepharose CL-4B affinity chromatography, according to Torres et al. [56].
Preparation of chitosan beads
Control chitosan beads were prepared by dissolving 4 % (w/v) pure chitosan in 5 % (v/v) acetic acid solution for 24 h at room temperature. Then, the opalescent brown-yellow solution was vacuum filtered and blown using a compressed air nozzle into a 0.1 M NaOH solution (chitosan:NaOH ratio of 1:10) to form coacervate beads under slow stirring (50 rpm) for 24 h at room temperature to complete the coagulation process. Beads were filtered and rinsed thoroughly with distilled and Milli-Q water, and then stored in refrigerator for further use. Particles 2.5 mm average diameter were obtained.
Chemical modification of chitosan before bead manufacturing
For the synthesis of modified beads, 4 % (w/v) d-lactose or 4 % (w/v) d-fructose was added to a freshly prepared 4 % (w/v) pure chitosan in 5 % (v/v) acetic acid solution and heated at 50 °C under stirring for 4 h. On the other hand, 1 % (w/v) APC, 10 % (w/v) TPP or 1 % (w/v) PVA were added to freshly prepared 4 % (w/v) pure chitosan in 5 % (v/v) acetic acid solution while vigorously stirring for 4 h at room temperature. PVA was prepared at 5 % (w/v) in absolute ethanol and 10 ml of the solution was added to chitosan suspension and immediately coagulated using a 1/10 mass/volume ratio. Furthermore, in all experiments, the modified chitosan beads were produced, washed and stored using the same production procedure as that of control beads [60].
Chemical surface modification of chitosan beads
Non-modified chitosan beads were mixed in 100 mM bicarbonate buffer (pH 10.05) in a mass/volume ratio of 1/10, and 1 % (v/v) TNBS or 1 % (v/v) DMF was added. The suspension was stirred at 50 rpm and heated at 55 °C for 6 h. Beads were filtered and rinsed thoroughly with an isopropanol-water mixture (1:1) for 30 s, followed by distilled water [10, 40].
Chitosan bead cross-linking with glyoxal
Chitosan beads were settled in 100 mM bicarbonate buffer (pH 10.05) in a mass/volume ratio of 1/10 and glyoxal was added up to a 3 % (v/v) final concentration. The suspension was magnetic stirred for 12 h at room temperature and the particles were filtered, soaked with distilled water and stored until further modification [62].
Natural or modified chitosan bead activation using glutaraldehyde
Screening assays with pure chitosan beads were performed at three different activation conditions: 1 % (v/v), 2 % (v/v) and 5 % (v/v) glutaraldehyde (GA) in 50 mM phosphate buffer (pH 7.0) using a mass/volume ratio of 1/5. They were incubated at room temperature for 20, 4 and 1 h, respectively. Immediately, supports were washed with distilled water, followed by activity buffer, being finally vacuum dried and weighed.
Next, for activation with glutaraldehyde, natural or modified chitosan beads (10 g) were suspended in 50 ml of 2 % (v/v) glutaraldehyde solution mixed with 50 mM phosphate buffer (pH 7.0) using a mass/volume ratio of 1/5. The suspension was stirred at 120 rpm for 4 h at room temperature. Then, beads were filtered and rinsed thoroughly with distilled water followed by Milli-Q water to remove the excess of the activating agent and immediately filtered and vacuum dried [20].
Chitosan derivatives were produced in triplicate using three different batches of chitosan particles. The experimental data shown are the average of the results where experimental data error was never above 5 %.
Natural or modified chitosan bead activation using glycidol
Glyceryl supports were prepared according to Adriano et al. [1] with several modifications. First, chitosan beads (10 g) were put into an ice bath under constant stirring and mixed with an aqueous solution containing 1.7 M NaOH (mass/volume ratio of 1/0.33) and 0.75 M NaBH4 (mass/volume ratio of 0.0136/1). Then, 0.48 ml of glycidol (0.75 M final concentration) was added gently and kept under mechanical agitation (80 rpm) for 18 h at room temperature, being finally washed until neutrality with distilled water. Glyoxyl derivatives were achieved by mixing beads in 2 ml of 0.1 M NaIO4 solution per gram (mass/volume ratio of 1/0.064) for 2 h at room temperature. Afterwards, glyoxyl supports were washed extensively with distilled and Milli-Q water, vacuum dried and stored at 4 °C until further use [58].
Natural or modified chitosan bead activation using epichlorohydrin
In the case of activation with epichlorohydrin (1-chloro-2,3-epoxypropane, Carlo Erba Analyticals, Rodano, Italy), vacuum-dried water-washed chitosan beads (10 g) were mixed with 10 ml of 2 M NaOH in ice bath until temperature reached about 0 °C. Then, 3 ml of 0.12 M freshly prepared NaBH4 solution was added, keeping pH in the range of 13-14. Immediately, 2 ml of epichlorohydrin per gram of chitosan beads was carefully added and the suspension was kept under magnetic stirring in ice bath for 2 h. Next, derivatives were withdrawn from ice bath and stored with agitation (150 rpm) until completion of 24 h at room temperature. Then, derivatives were extensively washed with distilled water until neutrality to remove the excess of epichlorohydrin and immediately vacuum dried. Afterwards, derivatives were suspended in 0.1 M NaIO4 prepared in 100 mM phosphate buffer (pH 7.0) for 2 h at room temperature with mechanical agitation, readily washed with Milli-Q water and vacuum dried [1].
Agarose bead activation with epichlorohydrin
First of all, 0.1 g of water-washed, vacuum-dried agarose CL-4B was mixed with an aqueous solution containing 2 M NaOH, 0.1 g NaBH4 and 1 ml of epichlorohydrin. After pH was verified to be around 13–14, suspension was incubated for 24 h at room temperature. Then, agarose was washed with distilled water until neutrality, suspended in 0.5 M NaOH solution and stored at 4 °C. The epoxy group title of activated agarose was 24 µmol of epoxy groups per g of wet gel.
Commercial pre-activated glyoxyl-agarose supports
Beads were deeply washed with water until pH 7.0 was reached and vacuum dried. Then, 1 g of drained gel was suspended in 8.50 ml of Milli-Q water and immediately 0.0306 g of NaIO4 was added. The suspension was kept under constant stirring for 2 h at room temperature and then supports were prepared for enzyme immobilization.
Determination of aldehyde group content on activated supports
The concentration of aldehyde groups in chitosan gel particles was calculated according to the methodology proposed by Adriano et al. [1].
Determination of oxirane group content on activated supports
Analysis of oxirane groups was performed according to Sundberg and Porath [54] with several modifications proposed by Torres et al. [56].
Optical and scanning electron (SEM) microscopy
An Olympus BH-2 (Tokyo, Japan) optical microscope equipped with a digital reflex camera system (Nikon D3100, Tokyo, Japan) was employed for the analysis of the macroscopic characteristics (size, shape, color) of the produced derivatives. On the other hand, for visualization of the surface morphology of the hybrid chitosan supports, scanning electron microscopy (SEM) was used. Samples were instantaneously frozen in liquid nitrogen and freeze dried using a Heto FD 2.5 equipment (Thermo Electron Corporation, Leicestershire, UK) under 5 × 10−3 mbar vacuum and −45 °C. When dried, samples were covered with a thin layer of gold (10 nm) using a combined metal/carbon sputter coater deposition system (Spi Supplies, 12157-AX, Structure Probe, Inc., West Chester, USA) operated in Argon atmosphere at 18 mA for 100 s and were examined using a JEOL JSM-35C (Jeol, Tokyo, Japan) scanning electron microscope with an acceleration tension of 20 kV and equipped with a SemAfore photograph acquisition system.
Multipoint covalent attachment of l-arabinose isomerase on chitosan supports activated with glutaraldehyde
Immediately after the activation process, enzyme immobilization was performed. For a 1 % (v/v) GA activation, the enzyme (10 mg of pure L-AI per gram of support) was added to the activated support (mass/volume ratio of 1/10) in sodium phosphate buffer with the addition of 0.6 M NaCl and was incubated for 24 h at room temperature with agitation. Then, for 2 % (v/v) GA, activated chitosan beads (1 g) were suspended in 10 ml of enzyme solution in 100 mM phosphate buffer (pH 7.0) or 100 mM sodium acetate buffer (pH 5.6) or 100 mM carbonate/bicarbonate buffer (pH 10.05) as immobilization buffers (10 mg of pure L-AI enzyme) under stirring (120 rpm) for 12 h at room temperature. The latter conditions were used only with the best biocatalysts for the evaluation of thermal stability at different pH; otherwise, assays were carried out at pH 7.0. For 5 % (v/v) GA, the enzyme in 50 mM phosphate buffer (pH 7.0) was incubated for 2 h at room temperature under gentle stirring. Insoluble biocatalysts were separated from supernatants by metal sieves (Retsch, Haan, Germany) and deeply washed with 500 mM phosphate buffer (pH 7.0) followed by 50 mM phosphate buffer (pH 7.0) and distilled water. Derivatives were then dried with filter paper, weighed and stored at 4 °C. Supernatants were filtered, clarified and tested for protein quantity and remaining L-AI activity. Derivatives incubated in pH 10.05 were reduced by addition of 0.1 % (w/v) NaBH4 under gentle stirring for 30 min at room temperature and extensively washed in sequence with distilled and Milli-Q water. Finally, as a presumptive analysis for enzyme inactivation under the immobilization conditions, a control experiment with the free enzyme was performed. In that way, incubation times were optimized according to the residual activity obtained in each activation condition.
Multipoint covalent attachment of l-arabinose isomerase on chitosan supports activated with epichlorohydrin
Epichlorohydrin pre-activated supports were poured in 50 ml of 100 mM phosphate buffer (pH 7.0) with the addition of 0.43 g of NaIO4 per gram of support and incubated for 2 h at room temperature. Then, particles were washed with distilled water, filtered and dried, mixed with a solution containing 10 mg of enzyme per gram of beads in 50 mM phosphate buffer (pH 7.0) and incubated at room temperature for 24 h with permanent agitation (120 rpm). Next, the insoluble derivatives were filtered and extensively washed with distilled water and, immediately, epoxy groups were blocked by reaction with 0.2 M β-mercaptoethanol for 1 h with magnetic stirring at room temperature. Afterwards, biocatalysts were washed with high ionic strength salt solution (1.5 M NaCl) in 50 mM sodium phosphate buffer (pH 7.0) for 12 h with mild stirring for removal of non-covalently bond enzyme, followed by distilled water. Schiff’s bases of derivatives were reduced by addition of 0.1 % (w/v) NaBH4 during 30 min at room temperature and were washed with distilled water, to remove the residual reducing agent, and Milli-Q water. Biocatalysts were filtered, rinsed in 50 mM phosphate buffer (pH 7.0), washed thoroughly with Milli-Q water and stored at 4 °C in a sterile bottle. Simultaneously, supernatants were recovered and assayed for protein concentration and residual L-AI activity.
Multipoint covalent attachment of l-arabinose isomerase on chitosan supports activated with glycidol
A mass of 10 mg of L-AI prepared in 10 ml of 0.1 M carbonate/bicarbonate buffer (pH 10.05) was added to 1 g of chitosan-glyoxyl activated support in a mass/volume ratio of 1/10. The preparation was kept under mild stirring at 25 °C for 36 h and, after that, supports were reduced by addition of 0.1 % (w/v) NaBH4 for 30 min at room temperature, followed by washing with distilled water and Milli-Q water [1, 55].
Glyoxyl-agarose l-arabinose isomerase immobilization
For activated agarose beads, a mass of 30 mg of L-AI enzyme was initially dissolved in 10 ml of 100 mM carbonate/bicarbonate buffer (pH 10.05). Depending on the degree of activation (low, high or very high) of commercial supports, pre-activated agarose beads (1 g) were suspended in 3.5, 7 or 10 ml, respectively, of enzyme solution under magnetic stirring (120 rpm) for 24 h at room temperature or until absorbance at 280 nm remained constant. Then, derivatives were filtered, thoroughly rinsed with 100 mM phosphate buffer (pH 7.0) for the elimination of the unbound protein and finally washed with distilled water. Then, Schiff’s bases were reduced using a 120 mM NaBH4 final concentration at 25 °C during 30 min under agitation and beads were rinsed with distilled water, followed by 50 mM phosphate buffer (pH 7.0) and 0.01 % (w/v) sodium azide. Finally, derivatives were vacuum dried and stored at 4 °C [36].
Epoxy-activated agarose l-arabinose immobilization
The epoxy-activated beads (1 g) were mixed with 1 mg of pure enzyme dissolved in 5.25 ml of 100 mM, sodium carbonate/bicarbonate buffer (pH 9.0) and incubated for 20 h under mild stirring at room temperature. Absorbance and activity controls were made during enzyme immobilization to ensure the covalent binding of the enzyme. Then, biocatalysts were washed twice for 15 min each with gentle stirring with one volume 0.9 M NaCl in 50 mM sodium phosphate buffer (pH 7.0), followed by 50 mM sodium phosphate buffer (pH 7.0). Finally, the remaining groups were blocked with 0.2 M β-mercaptoethanol for 2 h at room temperature, washed with 50 mM sodium phosphate buffer (pH 7.0), vacuum dried and stored at 4 °C until enzyme activity was determined [38].
l-arabinose isomerase immobilization in a methacrylate resin
Eupergit C250L®, which does not require previous activation, was used as methacrylate resin. For this purpose, 0.5 g of dry resin was suspended in 5.25 ml of 1 M potassium phosphate buffer (pH 7.5) with 1 mg of pure L-AI and incubated at room temperature under constant stirring for 24 h. Afterwards, biocatalyst was separated from supernatant and washed twice with one volume each of 1 M potassium phosphate buffer (pH 7.5), 0.1 M potassium phosphate buffer (pH 7.5) and 50 mM sodium phosphate buffer (pH 7.0) during 15 min each in permanent agitation at room temperature [7, 25]. Finally, the remaining groups were blocked with 0.2 M β-mercaptoethanol for 12 h at 4 °C, washed, vacuum dried and stored at 4 °C until enzyme activity assay.
Thermal stability assays
Both soluble and immobilized L-AI were incubated for 30 h at 60 °C in 50 mM sodium phosphate buffer (pH 7.0) with the addition of 0.3 mM MnCl2. At different time gaps, samples were withdrawn, put into an ice bath and residual activities were properly assessed according to the methodologies proposed above. In addition, thermal stability experimental results were adjusted according to the single-step non-first-order model proposed by Sadana and Henley [49].
Operational stability assays
Assays for d-galactose isomerization were done by pouring 0.5 g of vacuum dried immobilized L-AI derivative for 10 consecutive batch cycles. Reaction was performed in a closed bioreactor at 60 °C under magnetic stirring in a 2.5 ml total volume. After every cycle, derivatives were extensively washed with Milli-Q water to remove substrates and products, weighed and poured again in reactor for next cycle.
Protein determination
Protein was determined according to the methodology described by Bradford [9] using bovine serum albumin (BSA) as standard. When residual protein in supernatant was low, bicinchoninic acid (BCA, Pierce, Rockford, USA) methodology [59] was used instead.
l-arabinose isomerase activity assay
Immobilized L-AI biocatalysts assays were performed using the same enzyme/substrate ratio as that for the soluble enzyme. Reactants were added as follows: 1 g of insoluble biocatalyst, 17.15 ml of 50 mM phosphate buffer (pH 7.0), 0.35 ml of 100 mM MnCl2·4H2O prepared with 50 mM phosphate buffer (pH 7.0) solution and 17.5 ml of 1 M d-galactose prepared with 50 mM phosphate buffer (pH 7.0) solution, for a total volume of 35 ml. The reaction mixtures were placed in a thermostatic (Vicking, BA, Argentine) bath at 50 °C and 120 rpm for 1 h. Then, beakers were pulled out and instantly put on ice bath for 30 min to stop the isomerization reaction. Biocatalysts were separated by means of sieves and extensively washed with distilled water, vacuum dried and stored at 4 °C for time, thermal and operational stability assays. On the other hand, supernatants and the washes done to derivatives were collected, clarified through filtration and assayed, in triplicate for each sample, for protein quantity and enzyme activity. One unit of l-arabinose isomerase activity was defined as the quantity of enzyme that produced 1 µmol of d-tagatose per minute under assay conditions.
L-AI activity was determined by the amount of d-tagatose produced according to the cysteine-carbazole-sulfuric acid method proposed by Dische and Borenfreund [14]. All experiments were performed at least three times and the corresponding calibration curves were constructed employing d-tagatose as standard any time new reagents were prepared. Activity assessment was conducted in a slightly different way with Eupergit C250L and epoxy-activated agarose resins, where a final volume of 1 ml was used.
Immobilization parameters
Immobilized protein yield (η i ) was indirectly calculated as the amount of protein that disappeared from supernatant in relation to the protein quantity given initially to supports and to the protein lost after the washes performed to biocatalysts. Recovered activity yield (φ i ) of the immobilization process was calculated by dividing the apparent specific activity of the immobilized enzyme derivatives, A app, (U mg−1 g −1gel ) and the theoretically immobilized (U mg−1 g −1gel ) enzyme, because the offered enzyme load was known and the number of theoretically immobilized enzyme units per gram of gel can be calculated. These values were always in agreement with the reduced activity in the supernatant and in the washes performed to biocatalysts. The percentage of unbound enzyme was calculated dividing the remaining activity in the supernatant and washes by the enzyme activity measured in the blank, taking into account the dilution due to the addition of gel beads. Equivalent activity was calculated as the relation between the apparent specific activity of the immobilized enzyme derivatives, A app, (U mg−1 g −1gel ), and the effective specific activity bound to immobilized derivatives (U mg−1 g −1gel ). Blank assays were run with the soluble enzyme under the same conditions as those of immobilization assays and it was observed that L-AI always maintained 100 % activity. Finally, for the analysis of the quality and ligation efficiency of l-arabinose isomerase activity to supports, apparent enzyme activity A app, (U g −1gel ) was related to the protein quantity (in mg) incorporated to each biocatalyst. Stabilization factor was defined as the relation between the half-life of the immobilized derivate and the half-life of the soluble enzyme.
Results
Evaluation of l-arabinose isomerase immobilization process through a screening study on pure chitosan employing different activating agents
The obtained beads presented a spherical form with an average diameter of 2.5 mm. No significant differences were found in size or shape for all analyzed treatments. Moreover, the produced covalent bonds were confirmed by treating derivatives with a high ionic strength buffer for 12 h and 24 h under permanent stirring, followed by analysis of enzyme activity and protein quantity in all supernatants. In all cases, no enzyme was released to the solution, confirming the covalent nature of the interaction between enzyme and supports.
Native chitosan was tested with different activating agents for the assessment of the most adequate activating agent for L-AI immobilization.
Recovered activity with glycidol was lower (11 %) than that of the other agents. Furthermore, almost half of the offered enzyme remained in solution and was not bound to support, as seen in the protein and assays performed to supernatant. This could be partially explained by the high concentration of glyoxyl groups on the derivative, where reaction between two proximal glyoxyl groups could have been evidenced, leading to inactivation of several groups [46, 52]. The enzyme bound to support could have experimented inactivation due to the combined effect of pH and reaction time in enzyme immobilization. The latter effect could have produced several diffusional constraints and protein structure distortion, whereas at pH 10, l-arabinose isomerase presented no activity and, mostly, could have undergone an irreversible inactivation. Hence, after analysis of Table 1, glutaraldehyde and epichlorohydrin were selected as activating agents for their use in modified chitosan beads.
l-arabinose isomerase glutaraldehyde-activated chitosan biocatalysts
Initially, to test the best activation conditions for the enzyme, glutaraldehyde (GA) was used as activating agent in different concentrations for L-AI immobilization in non-modified chitosan beads. For these derivatives, immobilization yield (η i ) and recovery activity (φ i ) results are shown in Table 2.
Results showed that almost all offered enzyme (10 mg) was immobilized in 1 g of insoluble biocatalyst when using the three selected activation degrees. Activation with 2 % (v/v) GA showed the best stabilization factor for the enzyme in chitosan. The derivative with the highest activation degree could have partially inactivated the enzyme, leading to distortion of the protein structure when immobilized, because of the high reactivity of GA at high concentration. Furthermore, a high number of GA groups on activated chitosan could have led to an increased superficial hydrophobicity, which is not favorable for L-AI, an enzyme with hydrophilic characteristics. The 1 % (v/v) GA concentration could not have been sufficient to establish the number of covalent bonds necessary between support and enzyme, equivalent activity being then lower than that of the 2 % (v/v) GA-activated chitosan derivative. Therefore, 2 % (v/v) GA was selected as the adequate concentration of this activating agent for immobilization of L-AI on modified chitosan supports [2]. For this purpose, L-AI was immobilized on 22 modified chitosan derivatives and assayed for activity and protein bound to each support. Results of best derivatives using GA as activating agent are exhibited in Table 3.
After examining GA-activated chitosan beads under optical microscope, only native and d-fructose treated chitosan beads presented superficial rugosity and mesoporosity (see supporting information). In addition, both TNBS and DMF-treated chitosan beads showed a more compact surface although, when broken, similar internal porosity was observed in comparison with that of native chitosan particles. On the other hand, APC-treated and 3 % (w/v) alginate—2 % (w/v) chitosan particles showed an irregular and microporous surface whereas PVA-chitosan, 10 % (w/v) TPP—4 % (w/v) chitosan and glycerol-treated chitosan beads presented a denser area and a lesser microporosity in comparison with d-fructose derivative.
As seen in Table 3, immobilization yield (η i ) values were above 90 % for most analyzed biocatalysts. Chitosan-PVA derivative showed a higher apparent activity than control beads. This could be attributable to the additional contribution and higher interaction of hydroxyl groups by PVA, which allows glutaraldehyde to react mainly with amine groups though also, in a lesser extent, with hydroxyl groups. This would increase or, at least, maintain the binding capacity of the enzyme to modified supports [41].
Conversely, treatment of chitosan with TNBS and DMF did produce an increase in hydrophobicity through chemical surface modification of chitosan beads, as seen in the Rose Bengal dye assay (data not shown). However, a reduction of η i in DMF-modified chitosan derivatives would respond to a steric effect that interfered with the access of the enzyme to the activated matrix for covalent attachment. Besides, an increased hydrophobicity of the bead surface of both derivatives compared to that of control would contrast with the hydrophilic nature of the studied enzyme, resulting in low apparent activities. Furthermore, treatment of chitosan with TNBS or DMF reduced chitosan free amine groups, which consequently increased bead hydrophobicity. Hence, if this effect were the only probable cause for the low immobilization yield, it would be present on both derivatives, a situation that was not experimentally verified [40]. In addition, the stabilization effect of the enzyme in DMF-chitosan biocatalyst was attributable to the presence of certain hydrophobic interactions that counterbalanced the effect of chitosan -NH3 + groups.
On the contrary, better results of φ i and equivalent activity were achieved with the chitosan-TPP hydrogel. In this sense, although TPP reduced chitosan free amine groups, it provided two beneficial actions: firstly, the ionic gelation with TPP conferred to chitosan structure a better tridimensional reorganization, which allowed the generation of macroporous surfaces with subsequent higher stability than that of control [57]; secondly, interaction of TPP with chitosan brought the support that both a higher hydrophobicity and an increased water retention, which allowed a better stability of the enzyme in comparison with that of the untreated control [6].
The synthesis of alginate-chitosan complexes should stimulate the hydrogel structure due to the increase in the number of possible interactions between activating agent and support in comparison with pure chitosan beads. This behavior, though, was not observed. However, as expected, an increase in recovered activity was appreciated in PVA-chitosan beads.
Furthermore, alginate hydrogels are not recommended for reactions at temperatures above 60 °C, whereas chitosan is much more resistant even at temperatures up to 75 °C. Although the mixture of alginate-chitosan would improve this behavior, this was not evidenced experimentally [34]. Hence, the technological interest of this derivative and its application to isomerases is scarce.
For GA-activated chitosan beads, the N-alkylated derivative with d-fructose was the best in terms of recovered (63.56 %) and equivalent (68.59 %) activity after immobilization. N-alkylation of chitosan surface allowed a lesser distortion and better structural reorganization of immobilized enzyme structure while avoiding overactivation effects because d-fructose mainly acted as a space arm. This effect resulted in a higher activity per gram of the biocatalyst in relation with the other synthesized chitosan derivatives. Besides, the inclusion of d-fructose into chitosan matrix reduced the quantity of free amine group concentration which, as a direct effect, also reduced the metal chelating ability of chitosan [45]. Surprisingly, immobilized L-AI on d-lactose-chitosan derivative did not show any activity. First of all, it was previously proved that d-lactose bound tightly to chitosan structure through non-covalent interactions [19] and, as a consequence, amine groups were not altered. In that way, when support was activated with GA, free amine groups reacted with GA and, during L-AI immobilization course, d-lactose was still present in chitosan beads, thus causing enzyme inactivation through Maillard reaction. This behavior was clearly seen as the beads were progressively turning browner with time whereas, in freshly prepared beads, they still presented enzyme activity [17].
Finally, treatment of chitosan with glycerol, urea, sodium phosphate, ethanol and formaldehyde did not show any improvement in relation to untreated chitosan. In fact, several modifications did produce the binding of the enzyme to support (η i different from zero) although total enzyme inactivation. Furthermore, the use of glyoxal as cross-linking agent to increase the mechanical resistance of chitosan beads and consequently, enzyme stability and activity on the supports, did not produce the desirable effect. Hence, for an effective visualization of the desirable characteristics, glyoxal concentration should be higher and the treatments longer and more drastic than the employed ones.
l-arabinose isomerase epichlorohydrin-activated chitosan biocatalysts
The use of epichlorohydrin as activating agent revealed an η i between 56.7 and 99.7 % for all produced biocatalysts, which was always higher than that of control beads except for TNBS (56.7 %) and APC (75.5 %) supports. However, these values were overall low in comparison with glutaraldehyde-activated derivatives. Besides, recovered activities were extremely low when related with total activity.
The analysis of recovered activities proved them to be lower than that of the offered activity. However, equivalent activities of most biocatalysts are equal or slightly superior to those of control beads. In that way, the use of epichlorohydrin for L-AI immobilization to the modified chitosan hydrogel gave the enzyme a certain stability. Since this behavior was observed on all biocatalysts, this would indicate an overactivation effect which produced enzyme structural destabilization and inactivation. Consequently, shorter reaction times between enzyme and epichlorohydrin-activated chitosan beads would have been needed for an adequate L-AI immobilization. In addition, lower epichlorohydrin concentration would have been required for support activation to obtain derivatives with a lower density of epoxy groups [46, 55].
Thermal stability of soluble L-AI and best chitosan biocatalyst
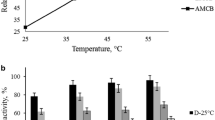
Thermal stability is a relevant property for the use of enzymes at industrial scale. In addition, the immobilization process generally improves the enzyme thermal stability and its efficient reutilization, which also depends on the selected immobilization strategy. Therefore, thermal stability of the best l-arabinose isomerase insoluble derivative, d-fructose-chitosan activated with 2 % (v/v) glutaraldehyde biocatalyst, was assessed at 60 °C and is shown in Fig. 1a.

Thermal stability of soluble L-AI and immobilized on d-fructose-chitosan support activated with 2 % (v/v) glutaraldehyde at 60 °C and pH 7.0 for 30 h. a Filled square soluble L-AI; chitosan derivatives: filled triangle pH 5.6; filled circle pH 7.0 and filled inverted triangle pH 10.05. Enzyme charge: 10 mg per gram of activated support (1800 U g−1). Initial activity was defined as 1. The bars are the standard deviations of triplicates. b Pentagon soluble L-AI; glyoxyl-agarose derivative: right angled triangle pH 10.05; Enzyme charge: 30 mg per gram of activated support (5400 U g−1). Initial activity was defined as 1. The bars are the standard deviations of triplicates
Results revealed that immobilization at pH 10.05, followed by pH 7.0 and pH 5.6, clearly originated the most stable derivative. Furthermore, at all assayed conditions, immobilized enzyme stability was higher than that of the soluble enzyme. There were more covalent bonds between enzyme and activated support at alkaline pH than at neutral or slightly acidic pH because ε-amine groups of l-lysine are more nucleophilic when not in the form of –NH3 +. Nonetheless, the best glyoxyl-agarose derivative (VHDG-6BCL) was also evaluated for thermal stability at an immobilization pH of 10.05, as seen in Fig. 1b.
Deactivation curves were fitted according to Sadana–Henley model. Inactivation constants, half-life times and stabilization factors for the three conditions tested are shown in Table 4.
As seen in Table 4, stability is highly favored as a consequence of the multiple covalent bonds achieved at pH 10.05 (211 times) in comparison with the derivatives achieved at pH 7.0 and 5.6. Since at neutral and acidic pH the nucleophilicity of l-lysine ε-amine groups is clearly reduced, so, there were less enzyme-activated support bonds than at alkaline pH. In addition, although glutaraldehyde is a very potent activating agent and would even act at acid pH, the bonding process is clearly favored at pH above pKa of the ε-amine groups of l-lysine. In the case of glyoxyl-activated agarose biocatalyst, a significant thermal stability was achieved, with a stabilization factor of 85, when compared to that of soluble enzyme. In both situations, stabilization was clearly improved through immobilization, taking into account the good thermal stability of soluble enzyme at the assayed temperature.
Operational stability of d-fructose chitosan derivative
l-arabinose isomerase immobilized on chitosan N-alkylated d-fructose derivative at pH 10.05 was used for the evaluation of the operational stability. Results are shown in Fig. 2.

Operational stability of L-AI immobilized on d-fructose-chitosan support activated with 2 % (v/v) glutaraldehyde at 60 °C and pH 7.0. Initial enzyme activity was defined as 100 %. The bars are the standard deviations of triplicates
The insoluble derivative was reused batchwise for 10 cycles for the isomerization of d-galactose to d-tagatose. After that, the residual activity, as relative activity, was more than 80 %. Hence, the enzyme was clearly stabilized after the application of the selected immobilization strategy.
l-arabinose isomerase immobilized on comercial resins
The enzyme was also immobilized on commercial supports because these matrixes have both an adequate morphological appearance and a controllable activation degree that guarantee a correct geometric congruence between enzyme and support, thus allowing different coupling possibilities between protein and support. Clearly, with the use of these resins, most properties and reaction conditions are well known in comparison with those of de novo produced materials.
Figure 3 shows the SEM microphotographs of the inner and outer structure of the best chitosan biocatalyst and the tested derivatives obtained with commercial resins.

Scanning electron microscopy (SEM) of best biocatalysts for the interior (a) and the surface (b) of L-AI immobilized on GA-activated d-fructose chitosan support; L-AI immobilized on glyoxyl-agarose matrix (c) and L-AI immobilized on Eupergit C250L (d) at different magnification ranges
Figure 3a shows the mesoporous structure of the obtained chitosan derivative when fractured, whereas Fig. 3b presents the surface area of the biocatalyst when L-AI was immobilized. Although treatment with d-fructose generated a clean surface, it did not change the inner chitosan structure, which remained quite uniform in pore size and shape after the chemical modification. This reinforced the idea that the treatments performed on chitosan modified only the bead surface, where the enzyme mainly interacts with support. Furthermore, glyoxyl-agarose derivative shows a slightly rough surface (Fig. 3c) in comparison with a smoother surface on Eupergit C250L matrix, as shown in Fig. 3d. This could be the reason why the agarose derivative presented a better equivalent activity in relation with the acrylic resin.
As can be seen in Table 5, pre-activated glyoxyl supports allowed the immobilization of almost all offered protein, although the recovered enzyme activity was not in agreement with protein yield. In addition, agarose cross-linking degree and porosity did not show a significant influence on the quality of the enzyme activity. Nevertheless, activation degree did affect η i and φ i . Accordingly, only HDG-4BCL and VHDG-6BCL derivatives presented an acceptable equivalent activity, which was higher than that of native chitosan control. On the other hand, all offered protein was immobilized on Eupergit C250L and epoxy-activated agarose. These resins stabilized enzyme activity, as seen in the equivalent activity results (82 % for Eupergit and 96 % for epoxy-activated agarose), although only 1 mg of protein was offered to these supports. These results would indicate not only that structural reorganization of L-AI on these supports was appropriate but also that stabilization of the enzyme was accomplished [7, 25].
Discussion
l-arabinose isomerase (EC 5.3.1.4) is an enzyme of industrial interest since, in vivo, it catalyzes the isomerization of l-arabinose to l-ribulose and, in vitro, can produce the reaction of d-galactose to d-tagatose, a relevant nutraceutic. Hence, its applicability could be optimized if L-AI were efficiently immobilized on a low-cost support to achieve a biocatalyst with high operational stability and catalytic efficiency. In this sense, chitosan (poly-N-acetyl glucosamine) became a material with excellent chemical characteristics for immobilization. However, due to its high chelating ability through its primary amine groups [45], the use of pure chitosan is not recommended for metalloenzymes where Mn2+ and Co2+ ions are essential for enzyme activity [42]. Then, conveniently modified chitosan can be used for effective support synthesis in metalloenzyme immobilization by multipoint covalent attachment.
In this research, 22 chitosan supports were synthetized following several experimental protocols. Results revealed that the recovered activity in chitosan derivatives (φ i ) was, in all cases, lower than the offered activity during the immobilization process. However, activation and immobilization were the same in all cases; if the main cause of enzyme activity loss were the direct action of activating agents over the enzyme, all derivatives should present similar φ i values, which was not the case. Therefore, differences between treatments could be explained by both a structural distortion produced by hydrogel overactivation when the enzyme is immobilized and metal cofactor chelation by means of chitosan structure.
To determine which biocatalyst was better than others in terms of protein quality and enzyme incorporated to derivatives, the equivalent activity per gram of biocatalyst and mg of immobilized enzyme was used (Fig. 4).

Best L-AI insoluble biocatalysts achieved after employing covalent attachment immobilization strategies with different matrixes
In that way, the chemical modification of chitosan free amine groups with d-fructose for 4 h at 50 °C and further bead formation followed by cross-linking with 2 % (v/v) glutaraldehyde was the best procedure in terms of both incorporated L-AI activity and enzyme stabilization when compared to DMF-chitosan and native chitosan control. Results showed an equivalent activity of 60.72 and 68.59 % for DMF and d-fructose chitosan derivatives, respectively, in comparison with a 5 % equivalent activity for control particles. However, apparent gel activity was 11.6 and 96.2 U g−1 for DMF and d-fructose chitosan derivatives, respectively. The latter value is clearly higher than that of several commercial supports in terms of incorporated protein and activity yield. Yet, the use of a hydrophilic support such as agarose, for enzyme immobilization, resulted in an insoluble biocatalyst with a good equivalent activity (96.34 %). The acrylic resin with epoxy groups also showed a good equivalent activity (81.5 %). In this sense, both derivatives introduced minimal chemical modifications to enzyme whereas establishing strong secondary amine-carbon covalent bonds and a more hydrophobic microenvironment [7].
For glyoxyl-agarose derivatives, almost all offered protein was immobilized at the different activation degrees assayed. Hence, the higher the activation degree, the higher the enzyme activity that has been retained in the support. However, the hydrophobic alteration of the bead surface produced by reaction of agarose hydroxyl groups was not the most suitable microenvironment for L-AI, a clearly hydrophilic enzyme. For that reason, the relation between offered and immobilized enzymes was not linear as the activation degree was increased. In addition, these derivatives could immobilize higher quantities of enzyme than Eupergit C250L and epoxy-activated agarose CL-6B because only 1 mg of total protein was incorporated to the latter ones. Hence, the equivalent activity per mg of protein of these supports was lower than that of the best chitosan biocatalyst.
On the other hand, a commercial glyoxyl pre-activated support (VHDG-6BCL) with a threefold higher (28.9 mg) enzyme load than the best chitosan biocatalyst (9.3 mg) achieved an enzyme activity only 60 % higher than d-fructose-chitosan biocatalyst. In this sense, the high activation degree of the commercial resin could have led to partial enzyme denaturation due to a distortion of the tridimensional structure as a consequence of the number of covalent bondings between enzyme and support. Furthermore, the apparent enzyme activity per mg of protein of d-fructose chitosan derivative was 96.2 U mg g −1gel , whereas that of VHDG glyoxyl-agarose support was 161.80 U mg g −1gel . Consequently, the latter activity was higher than that of chitosan support because a higher protein quantity was immobilized. However, only 35.46 % of total activity remained active, which implied a huge loss of activity (18 mg of pure enzyme). In that way, only 32.1 % of the offered enzyme was incorporated, in contrast with the chitosan derivative, which immobilized 63.56 % of total activity, representing 6.6 mg of the initially applied 10 mg of pure enzyme. Hence, in this aspect, chitosan biocatalyst is better than VHDG derivative because a smaller quantity of the enzyme was lost. Furthermore, since glyoxyl-agarose resin has a higher cost the production of derivatives with low-cost support, such as chitosan, represents an economical advantage. Therefore, similar or better results were obtained with chitosan hydrogels than with commercial matrixes of controlled size and porosity, such as agarose or Eupergit C250L.
The greatest—although scarce—contributions in relation to l-arabinose isomerase immobilization were given by Zhang et al. [63] who, for Bacillus licheniformis L-AI, achieved an immobilization yield of 83.7 and 35.6 % and an activity recovery of 93.3 and 75.6 % for Eupergit C and CNBr-activated agarose supports, respectively, for covalent immobilization of L-AI (4 mg per gram of support). Even though Eupergit C immobilization process was subsequently optimized, results were not significantly improved. These results are in agreement with ours although immobilization yield values were slightly higher for the present research. Recovered activities, however, were about the same range when increasing the offered enzyme concentration.
They have also reported the only previous research where chitosan was used for the L-AI immobilization from B. licheniformis [64]. Herein, the enzyme was trapped non-covalently by adsorption interactions in 3 % (w/v) chitosan/0.136 mM TPP beads. An immobilization efficiency of 35.4 % and a recovered activity of 35.4 % were obtained, which were low in comparison with those of the produced chitosan-TPP derivative (94 % of immobilization yield and 23 % of recovered activity, respectively). However, the latter derivative had an apparent gel activity of 32 %, as a consequence of the enzyme-support covalent approach and subsequent better stability parameters. Furthermore, Geobacillus stearothermophilus L-AI expressed in Bacillus subtilis was immobilized onto 2 mg of chitopearls (commercial pre-activated chitosan beads) due to the higher thermal resistance of the support and a 43 % conversion yield after 62 h at 60 °C was attained [12]. However, since many characteristics of these particles and the immobilization conditions employed are unknown, results are difficult to be discussed in relation to the enzyme behavior on chitosan.
Several researches have reported the use of different organic matrixes—such as sodium alginate [26, 27, 32, 44, 65], mesoporous aminopropyl glass [64], polyethylenimine [24], agarose [29] and CNBr-agarose for L-AI immobilization and production of d-tagatose [43]. However, these researches have not gone much deeper into the understanding of the enzyme immobilization process or have explored the possibilities of chitosan for the achievement of stable enzyme biocatalysts.
Thermal stability assays revealed significant stabilization factors using the best chitosan derivative and glyoxal-agarose biocatalyst. In that way, covalent immobilization through multipoint attachment became an adequate approach for l-arabinose isomerase immobilization, which resulted in an increased enzyme stability and activity.
On the other hand, the analysis of operational stability showed that L-AI d-fructose chitosan derivative kept the activity above 80 % after 10 recirculation cycles at 60 °C whereas Rhimi et al. [44] reported activity results above 70 % after 8 cycles with Bacillus stearothermophilus US100 L-AI entrapped in 6 % (w/v) of sodium alginate on BaCl2 beads. Furthermore, Oh et al. [27] used particles produced with 4 % (w/v) sodium alginate and treated with 0.2 (v/v) glutaraldehyde, which gave good results for 8 days (or cycles), although the activity of the biocatalyst rapidly decreased.
In this research, a contribution for the study of l-arabinose isomerase from E. faecium DBFIQ E36 immobilization process was intended. Several modified chitosan derivatives achieved significant results such as good apparent gel activity, thermal resistance, operational stability, low swelling factor and increased mechanical resistance. In addition, stabilization factors for several chitosan biocatalysts were near to those of the best commercial derivatives.
The use of chitosan for matrix synthesis and further modification turned out to be a viable low-cost strategy for achieving highly active and stable L-AI insolubilized derivatives in comparison with high-cost commercial supports. In that way, the synthesis and use of supports adapted for L-AI immobilization became a topic of interest due to the current technological potential of d-galactose to d-tagatose bioconversion.
Finally, although the production of d-tagatose through a biological approach using l-arabinose isomerase proved profitable, for feasible industrial application the design of highly stable catalytic derivatives becomes mandatory. Therefore, the present research provided an increased knowledge for multimeric metalloenzyme immobilization such as l-arabinose isomerase. The goal was reached through the modification of chitosan biopolymer to obtain alternative beads adapted for a lower metal chelating ability, followed by immobilization to supports via multipoint covalent attachment strategy for achieving increased enzyme stability due to the multimeric nature of the enzyme.
References
Adriano WS, Mendonça DB, Rodrigues DS, Mammarella EJ, Giordano RLC (2008) Improving the properties of chitosan as support for the covalent multipoint immobilization of chymotrypsin. Biomacromolecules 9:2170–2179
Barbosa O, Ortiz C, Berenguer-Murcia A, Torres R, Rodrigues RC, Fernandez-Lafuente R (2014) Glutaraldehyde in bio-catalysts design: a useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv 4:1583–1600
Barbosa O, Torres R, Ortiz C, Berenguer-Murcia A, Rodrigues RC, Fernandez-Lafuente R (2013) Heterofunctional supports in enzyme immobilization: from traditional immobilization protocols to opportunities in tuning enzyme properties. Biomacromolecules 14:2433–2462
Berger J, Reist M, Mayer JM, Felt O, Gurny R (2004) Structure and interactions in chitosan hydrogels formed by complexation or aggregation for biomedical applications. Eur J Pharm Biopharm 57:35–52
Berger J, Reist M, Mayer JM, Felt O, Peppas NA, Gurny R (2004) Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur J Pharm Biopharm 57:19–34
Bhumkar DR, Pokharkar VB (2006) Studies on effect of pH on cross-linking of chitosan with sodium tripolyphosphate: a technical note. AAPS PharmSciTech 7:138–143
Boller T, Meier C, Menzler S (2002) Eupergit oxirane acrylic beads: how to make enzymes fit for biocatalysis. Org Proc Res Dev 6:509–519
Boudebbouze S, Maguin E, Rhimi M (2011) Bacterial l-arabinose isomerases: industrial application for d-tagatose production. Recent Pat DNA Gene Seq 5:194–201
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brady D, Jordaan J (2009) Advances in enzyme immobilization. Biotechnol Lett 31:1639–1650
Cantone S, Ferrario V, Corici L, Ebert C, Fattor D, Spizzo P, Gardossi L (2013) Efficient immobilisation of industrial biocatalysts: criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem Soc Rev 42:6262–6276
Choon J, Kim SB, Park SW, Han JK, Kim P (2008) Comparative analysis of tagatose productivity of immobilized l-arabinose isomerase expressed in Escherichia coli and Bacillus subtilis. Food Sci Biotechnol 17:655–658
DiCosimo R, McAuliffe J, Poulose AJ, Bohlmann G (2013) Industrial use of immobilized enzymes. Chem Soc Rev 42:6437–6474
Dische Z, Borenfreund E (1951) A new spectrophotometric method for the detection and determination of keto sugars and trioses. J Biol Chem 192:583–587
Donati I, Stredanska S, Silvestrini G, Vetere A, Marcon P, Marsich E, Mozetic P, Gamini A, Paoletti S, Vittur F (2005) The aggregation of pig articular chondrocyte and synthesis of extracellular matrix by a lactose-modified chitosan. Biomaterials 26:987–998
Fernandez-Lafuente R (2009) Stabilization of multimeric enzymes: strategies to prevent subunit dissociation. Enzyme Microb Technol 45:405–418
García-Bermejo AB, Cardelle-Cobas A, Ruiz-Mature AI, Montanes F, Olano A, Corzo N (2012) Effect of drying methods on the reactivity of chitosan towards Maillard reaction. Food Hydrocoll 29:27–34
Garcia-Galan C, Berenguer-Murcia A, Fernandez-Lafuente R, Rodrigues RC (2011) Potential of different enzyme immobilization strategies to improve enzyme performance. Adv Synth Catal 353:2885–2904
Hall LD, Yalpani MD (1980) Formation of branched-chain, soluble polysaccharides from chitosan. J Chem Soc Chem Commun 38:1153–1154
He P, Davi SS, Illum L (1999) Chitosan microspheres prepared by spray drying. Int J Pharm 187:53–65
Hernandez K, Fernandez-Lafuente R (2011) Control of protein immobilization: coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb Technol 48:107–122
Hwang ET, Gu MB (2013) Enzyme stabilization by nano/microsized hybrid materials. Eng Life Sci 13:49–61
Iyer PV, Ananthanarayan L (2008) Enzyme stability and stabilization-aqueous and non-aqueous environment. Process Biochem 43:1019–1032
Jørgensen F, Hansen OC, Stougaard P (2004) Enzymatic conversion of d-galactose to d-tagatose: heterologous expression and characterisation of a thermostable l-arabinose isomerase from Thermoanaerobacter mathranii. Appl Microbiol Biotechnol 64:816–822
Katchalski-Katzir E, Kraemer DM (2000) Eupergit® C, a carrier for immobilization of enzymes of industrial potential. J Mol Catal B Enzym 10:157–176
Kato Y, Onishi H, Machida Y (2001) Biological characteristics of lactosaminated N-succinyl-chitosan as a liver-specific drug carrier in mice. J Control Release 70:295–307
Kim HJ, Ryu SA, Kim P, Oh DK (2003) A feasible enzymatic process for d-tagatose production by an immobilized thermostable l-arabinose isomerase in a packed-bed bioreactor. Biotechnol Prog 19:400–404
Kim JW, Kim YW, Roh HJ, Kim HY, Cha JH, Park KH, Park CS (2003) Production of tagatose by a recombinant thermostable l-arabinose isomerase from Thermus sp. IM6501. Biotechnol Lett 25:963–967
Kim P, Yoon SH, Roh HJ, Choi JH (2001) High production of d-tagatose, a potential sugar substitute, using immobilized l-arabinose isomerase. Biotechnol Prog 17:208–210
Krajewska B (2004) Application of chitin-and chitosan-based materials for enzyme immobilizations: a review. Enzyme Microb Technol 35:126–139
Kumar MN, Muzzarelli RA, Muzzarelli C (2004) Chitosan chemistry and pharmaceutical perspectives. Chem Rev 104:6017–6084
Liang M, Chen M, Liu X, Zhai Y, Liu X, Zhang H, Xiao M, Wang P (2012) Bioconversion of d-galactose to d-tagatose: continuous packed bed reaction with an immobilized thermostable l-arabinose isomerase and efficient purification by selective microbial degradation. Appl Microbiol Biotechnol 93:1469–1474
Liese A, Hilterhaus L (2013) Evaluation of immobilized enzymes for industrial applications. Chem Soc Rev 42:6236–6249
Lim BC, Kim HJ, Oh DK (2008) Tagatose production with pH control in a stirred tank reactor containing immobilized l-arabinose isomerase from Thermotoga neapolitana. Appl Biochem Biotechnol 149:245–253
Maghami GA, Roberts GAF (1988) Studies on the adsorption of anionic dyes on chitosan. Macromol Chem Phys 189:2239–2243
Manrich A, Galvão CMA, Jesus CDF, Giordano RC, Giordano RLC (2008) Immobilization of trypsin on chitosan gels: use of different activation protocols and comparison with other supports. Int J Biol Macromol 43:54–61
Manzo RM, Simonetta AC, Rubiolo AC, Mammarella EJ (2013) Screening and selection of wild strains for l-arabinose isomerase production. Braz J Chem Eng 30:711–720
Mateo C, Bolivar JM, Godoy CA, Rocha-Martin J, Pessela BCC, Curiel JA, Muñoz R, Guisan JM, Fernández-Lorente G (2010) Improvement of enzyme properties with a two-step immobilization process on novel heterofunctional supports. Biomacromolecules 11:3112–3117
Mateo C, Palomo JM, Fernández-Lorente G, Guisán JM, Fernández-Lafuente R (2007) Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb Technol 40:1451–1463
Mendes AA, de Castro HF, Rodrigues DS, Adriano WS, Tardioli PW, Mammarella EJ, Giordano RC, Giordano RLC (2011) Multipoint covalent immobilization of lipase on chitosan hybrid hydrogels: influence of the polyelectrolyte complex type and chemical modification on the catalytic properties of the biocatalysts. J Ind Microbiol Biotechnol 38:1055–1066
Mucha M, Ludwiczak S, Kawinska M (2005) Kinetics of water sorption by chitosan and its blends with poly(vinyl alcohol). Carbohydr Polym 62:42–49
Oh DK (2007) Tagatose: properties, applications, and biotechnological processes. Appl Microbiol Biotechnol 76:1–8
Oh DK, Kim HJ, Ryu SA, Rho HJ, Kim P (2001) Development of an immobilization method of l-arabinose isomerase for industrial production of tagatose. Biotechnol Lett 23:1859–1862
Rhimi M, Messaoud EB, Borgi MA, Khadra KB, Bejar S (2007) Co-expression of l-arabinose isomerase and d-glucose isomerase in E. coli and development of an efficient process producing simultaneously d-tagatose and d-fructose. Enzyme Microb Technol 40:1531–1537
Rinaudo M (2006) Chitin and chitosan: properties and Applications. Prog Polym Sci 31:603–632
Rodrigues DS, Mendes AS, Adriano WS, Gonҫalves LRB, Giordano RLC (2008) Multipoint covalent immobilization of microbial lipase on chitosan and agarose activated by different methods. J Mol Catal B Enzym 51:100–109
Rodrigues RC, Ortiz C, Berenguer-Murcia A, Torres R, Fernández-Lafuente R (2013) Modifying enzyme activity and selectivity by immobilization. Chem Soc Rev 42:6290–6307
Ruhsing Pan J, Huang C, Chen S, Chung YC (1999) Evaluation of a modified chitosan biopolymer for coagulation of colloidal particles. Colloids Surf A Physicochem Eng Asp 147:159–364
Sadana A, Henley JP (1987) Single-step unimolecular non-first-order enzyme deactivation kinetics. Biotechnol Bioeng 30:717–723
Sashiwa H, Aiba SI (2004) Chemically modified chitin and chitosan as biomaterials. Prog Polym Sci 29:887–908
Sheldon RA, Van Pelt S (2013) Enzyme immobilisation in biocatalysis: why, what and how. Chem Soc Rev 42:6223–6235
Silva JA, Macedo GP, Rodrigues DS, Giordano RLC, Gonҫalves LRB (2012) Immobilization of Candida antarctica lipase B by covalent attachment on chitosan-based hydrogels using different support activation strategies. Biochem Eng J 60:16–24
Stepankova V, Bidmanova S, Koudelakova T, Prokop Z, Chaloupkova R, Damborsky J (2013) Strategies for stabilization of enzymes in organic solvents. ACS Catal 3:2823–2836
Sundberg L, Porath J (1974) Preparation of adsorbents for biospecific affinity chromatography. Attachment of group-containing ligands to insoluble polymers by means of bifuctional oxiranes. J Chromatogr 90:87–98
Tardioli PW, Fernández-LaFuente R, Guisán JM, Giordano RLC (2003) Design of new immobilized stabilized carboxypeptidase A derivative for production of aromatic free hydrolysates of proteins. Biotechnol Prog 19:565–574
Torres PR, Manzo RM, Rubiolo AC, Batista-Viera FD, Mammarella EJ (2014) Purification of an l-arabinose isomerase from Enterococcus faecium DBFIQ E36 employing a biospecific affinity strategy. J Mol Catal B Enzym 102:99–105
Vasconcellos FC, Goulart GAS, Beppu MM (2011) Production and characterization of chitosan microparticles containing papain for controlled release applications. Powder Technol 205:65–70
Vold IMN, Christensen BE (2005) Periodate oxidation of chitosans with different chemical compositions. Carbohydr Res 340:679–684
Walker JM (2002) The Bicinchoninic acid (BCA) assay for protein quantitation. In: Walker JM (ed) The protein protocols handbook, 2nd edn. Humana Press Inc., Totowa, pp 11–14
Xu Y, Du Y (2003) Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int J Pharm 250:215–226
Yalpani M, Hall LD (1984) Some chemical and analytical aspects of polysaccharide modifications. III. Formation of branched-chain, soluble chitosan derivatives. Macromolecules 17:272–281
Yang Q, Dou F, Liang B, Shen Q (2005) Studies of cross-linking reaction on chitosan fiber with glyoxal. Carbohydr Polym 59:205–210
Zhang Q, Chen C (2015) Direct acylation of aryl amines using dimethylformamide and dimethylacetamide as the acyl resources. J Saudi Chem Soc (in press)
Zhang YW, Jeya M, Lee JK (2011) Enhanced activity and stability of l-arabinose isomerase by immobilization on aminopropyl glass. Appl Microbiol Biotechnol 89:1435–1442
Zhang YW, Prabhu P, Lee JK (2009) Immobilization of Bacillus licheniformis l-arabinose isomerase for semi-continuous l-ribulose production. Biosci Biotechnol Biochem 73:2234–2239
Acknowledgments
This work was partially sponsored by funds of the projects CAI+D 2011 501 201101 00357 LI (Universidad Nacional del Litoral, Santa Fe, Argentina), PICT Bicentenario 2010 No. 0080 (ANPCyT, Buenos Aires, Argentina) and Argentina–Brazil Bilateral Cooperation Program BR/12/06 MINCyT-CAPES 2012 (Buenos Aires, Argentina). The authors declare no competing financial interest. The authors would like to thank the financial support of the Brazilian Research Agencies CNPq, CAPES, FINEP, FUNCAP and FAPESP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manzo, R.M., de Sousa, M., Fenoglio, C.L. et al. Chemical improvement of chitosan-modified beads for the immobilization of Enterococcus faecium DBFIQ E36 l-arabinose isomerase through multipoint covalent attachment approach. J Ind Microbiol Biotechnol 42, 1325–1340 (2015). https://doi.org/10.1007/s10295-015-1662-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-015-1662-1