
Abstract
The aim of this study was to evaluate the effect of low-level laser irradiation (LLLI) on the proliferation and viability of stem cells from human exfoliated deciduous teeth (SHED). Cells were irradiated or not (control) with an InGaAlP laser diode (660 nm, 30 mW, continuous action mode) using two different energy densities (0.5 J/cm2—16 s; 1.0 J/cm2—33 s). Irradiation was performed at 0 and 48 h, with the laser probe fixed at a distance of 0.5 cm from the cells. Cell proliferation was analyzed at 0, 24, 48, and 72 h by the Trypan blue exclusion method and MTT assay. Cell cycle and Ki67 expression were analyzed by flow cytometry. Apoptosis-related events were evaluated by expression of annexin V/PI and nuclear morphological changes by staining with DAPI. Differences between groups at each time were analyzed by the Kruskal–Wallis and Mann–Whitney tests, adopting a level of significance of 5% (p < 0.05). The results showed that an energy density of 1.0 J/cm2 promoted an increase in cell proliferation at 48 and 72 h compared to the control and 0.5 J/cm2 groups. Cell cycle analysis revealed a predominance of cells in the S and G2/M phases in the irradiated groups. This finding was confirmed by the increased expression of Ki67. Low positive staining for annexin V and PI was observed in all groups, and no nuclear changes were detected, indicating that cell viability was not affected by the energy densities tested. It can be concluded that the LLLI parameters used (660 nm, 30 mW, 1.0 J/cm2) promote the proliferation of SHEDs and the maintenance of cell viability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stem cells from human exfoliated deciduous teeth (SHEDs) were isolated by Miura et al. [1] as a population of clonogenic cells with proliferative capacity that are able to differentiate into a variety of cell types. Experiments using these cells have shown significant differences in the biology of stem cells obtained from deciduous and permanent teeth. When compared to stem cells isolated from the pulp of permanent teeth, SHEDs exhibited a higher proliferation rate [2] and a larger number of cell divisions—which could facilitate their in vitro expansion, as well as greater colony formation, in vivo osteoinductive capacity, and formation of different tissues of the dentin pulp complex [1].
The isolation of SHEDs is a simple and convenient process that causes little or no trauma considering that every child loses deciduous teeth, thus representing the perfect opportunity of recovering and storing stem cells to treat diseases or injuries in the future [3]. However, these cells need to be stored for subsequent clinical use, and their yield in culture is usually low. Thus, stimulation of in vitro proliferation is fundamental to obtain an adequate number of cells [4].
Low-level laser irradiation (LLLI) could be an effective strategy for promoting significant expansion of the number of cells in vitro and their subsequent differentiation and may therefore be a useful tool in in vivo regenerative processes and tissue engineering protocols. The important physiological effect of LLLI has been related to its capacity to stimulate the proliferation of different cell types [5]. Studies have shown that stimulation with LLLI increases the proliferation rate of different cells such as fibroblasts [6, 7], endothelial cells [8], and osteoblasts [9].
Few of the various sources of stem cells have been submitted to laser irradiation [10,11,12], a fact highlighting the need for further studies to better understand the effect of laser therapy on the proliferation of these cells. With respect to laser therapy applied to stem cells of dental origin, most studies have used stem cells derived from the pulp of permanent teeth, while investigations using other dental or periodontal sources of stem cells are sparse in the literature [12, 13]. Therefore, we tested the hypothesis that LLLI promotes proliferation of SHEDs. For this purpose, we performed in vitro cell proliferation and viability assays and evaluated cell cycle, apoptosis, and nuclear morphology in order to assess the influence of LLLI on the biological activity of SHEDs.
Materials and methods
The study was approved by the Ethics Committee of the Federal University of Rio Grande do Norte (Approval No. 34913314.4.0000.5537). Three human deciduous teeth exhibiting at least one third of physiological root resorption were used for the isolation of SHEDs.
Cell culture
After extraction, each tooth was immediately transferred to a Falcon tube (TPP, Trasadingen, Switzerland) containing 5 mL alpha-MEM medium (Gibco, Carlsbad, CA, USA) and stored at 4 °C. In a laminar flow chamber, the teeth were submitted to the protocol described by Vasconcelos et al. [14] to prevent possible contamination with microorganisms: three washes of 10 min each with a solution containing alpha-MEM medium supplemented with 10,000 IU/mL penicillin, 10 mg/mL streptomycin, 100 mg/mL gentamicin, and 250 μg/mL amphotericin B (all reagents from Gibco, Carlsbad, CA, USA).
SHEDs were obtained according to the protocol of Ginani et al. [15]. The pulp tissue was carefully removed by curettage using an endodontic excavator (Dentsply Maillefer, Ballaigues, Switzerland), and the extract was digested with 3 mg/mL collagenase I (Gibco, Carlsbad, CA, USA) and 4 mg/mL dispase (Gibco, Carlsbad, CA, USA) for 1 h at 37 °C. The cultures were incubated in alpha-MEM medium supplemented with 15% fetal bovine serum (Gibco, Carlsbad, CA, USA) at 37 °C in a 5% CO2 atmosphere until they reached 70–90% confluence. The medium was changed every 3 days.
In the first passage (P1), the cells were analyzed to confirm their stem cell nature according to the criteria proposed by Dominici et al. [16]. Briefly, an aliquot of cells was evaluated by flow cytometry using the Human MSC Analysis Kit (BD Stemflow™, BD Biosciences, San Jose, CA, USA), which permits to identify mesenchymal stem cells by detecting positive (CD105 PerCP-Cy5.5, CD73 APC, CD90 FITC) and negative (CD45, CD34, CD11b, CD19, HLA-DR PE) expression of cell surface markers. In addition, the cells were cultured in osteogenic and adipogenic differentiation medium (StemPro® Differentiation Kits, Invitrogen, Carlsbad, CA, USA) for up to 21 days, and the multipotent nature of SHEDs was confirmed by von Kossa (Abcam, Cambridge, UK) and Oil Red (Sigma-Aldrich, St. Louis, MO, USA) staining.
Experimental design
Figure 1 illustrates the steps performed in this study. After characterization, SHEDs were expanded and divided into three groups according to treatment: (1) control: no irradiation, (2) Laser 0.5: cells irradiated with a dose of 0.5 J/cm2, and (3) Laser 1.0: cells irradiated with a dose of 1.0 J/cm2. Next, assays were conducted for evaluating cell viability and proliferative capacity in all groups and to identify apoptosis-related and cell cycle events.

Study design. P passage, T time of analysis after the first irradiation (in hours)
Laser irradiation
Cells of groups 2 and 3 in the third and fourth passage (P3 and P4) were irradiated with an InGaAlP laser diode (Bio Wave LLLT Dual, Kondortech, São Carlos, SP, Brazil) in the continuous action mode using the following parameters: power of 30 mW, wavelength of 660 nm, doses of 0.5 and 1.0 J/cm2, irradiation time of 16 s (0.5 J/cm2) and 33 s (1.0 J/cm2), and tip diameter of 0.01 cm2. The irradiation probe was applied perpendicular to the plate at a distance of 0.5 cm from the cells. The power of the laser was measured with a LaserCheck Power Meter (Coherent Inc., Santa Clara, CA, USA) before each application.
The cells were irradiated at 0 and 48 h, and the analyses were carried out at 0, 24, 48, and 72 h after the first laser application. For the Trypan blue exclusion test and MTT assay, the cells were cultured in 24 and 96-well plates, respectively. Irradiation was applied to the center of each well. For the cell cycle, Ki67 expression, and apoptosis assays, the cells were cultured in 6-well plates and the laser was applied at five fixed and distinct points in each well. The cells were plated in such a way that there was an empty well between seeded wells to prevent the unintentional spread of light between wells during laser application.
Analysis of cell proliferation
Cell proliferation was analyzed by the Trypan blue exclusion method and by the MTT assay at 0, 24, 48, and 72 h after the first irradiation. All assays were carried out in quadruplicate (n = 4). For Trypan blue staining (Gibco, Carlsbad, CA, USA), the cells were cultured in 24-well plates at a density of 2 × 104 cells/well. The number of Trypan blue-stained cells in each well was counted in a Neubauer chamber (Optik Labor, Lancing, UK) at the time points analyzed. For the MTT assay (Invitrogen, Carlsbad, CA, USA), the cells were cultured in 96-well plates at a density of 5 × 103 cells/well and absorbance was measured in a microplate reader (Epoch, BioTek Instruments, Winooski, VT, USA) at 570 nm.
Cell cycle analysis
To evaluate the effects of laser therapy on the cell cycle, cells of each group were cultured in triplicate (n = 3) in 6-well plates at a density of 2 × 105 cells/well. The cells were processed after the intervals studied (0, 24, 48, and 72 h), and 5 μL propidium iodide (PI, 25 mg/mL; Invitrogen, Carlsbad, CA, USA) in 200 μL ice-cold PBS (Gibco, Carlsbad, CA, USA) was added for analysis in a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA, USA).
Analysis of Ki67 expression
The expression of nuclear protein Ki67 was analyzed by flow cytometry. For this purpose, the cells were cultured in triplicate (n = 3) in 6-well plates at a density of 2 × 105 cells/well. After 72 h (T72), the cells were processed and incubated with 10 μL of FITC-conjugated anti-Ki67 monoclonal antibody (Clone 7B11, Invitrogen, Carlsbad, CA, USA) for 20 min in the dark and analyzed in a flow cytometer (FACS Canto II, BD Biosciences, San Jose, CA, USA).
Analysis of apoptosis
The effects of laser therapy on apoptosis were analyzed using the FITC/Annexin V Dead Cell Apoptosis Kit with FITC annexin and PI for flow cytometry (Invitrogen, Carlsbad, CA, USA). The cells were cultured in triplicate in 6-well plates at a density of 2 × 105 cells/well. After the last interval analyzed (T72), the cells were processed and incubated with 3 μL FITC annexin V and 1 μL PI (100 μg/mL) for 15 min at room temperature in the dark. After incubation, the cells were analyzed by flow cytometry (FACS Canto II, BD Biosciences, San Jose, CA, USA), measuring fluorescence emission at 530 and 575 nm.
Analysis of nuclear morphology
To evaluate the presence of nuclear damage in the cells submitted to laser therapy, the cells were cultured in 24-well plates at a density of 2 × 104 cells/well. After 72 h, the cells were incubated with 4′-6-diamidino-2-phenylindole (DAPI; Molecular Probes, Carlsbad, CA, USA) diluted 1:1000 for 20 min in a dark chamber. The cells were examined under a fluorescence microscope (Eclipse Ti-U, Nikon Instruments Inc., Melville, NY, USA) to identify the presence of pyknotic nuclei and nuclear fragmentation in cells of the three groups.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA). Since data did not show normal distribution (Shapiro–Wilk test, p < 0.05), non-parametric analyses were performed. Differences between groups at each time point (0, 24, 48, and 72 h) were analyzed by the Kruskal–Wallis and Mann–Whitney tests, adopting a level of significance of 5% (p < 0.05; beta 0.2; power 0.8).
Results
Pulp cells were positive for the surface markers CD90 (98.8%), CD73 (99.9%), and CD105 (97.4%), while less than 1% of cells were positive for CD45, CD34, CD11b, CD19, and HLA-DR. Light microscopy revealed the deposition of mineralized matrix stained with von Kossa stain and the presence of lipid vesicles stained with Oil Red O, indicating the differentiation of cells into osteoblasts and adipose cells, respectively (Fig. 2).

a–d Expression of stem cell markers in SHEDs: a CD90, b CD73, c CD105, and d negative cocktail. e–f Photomicrographs of SHEDs subjected to osteogenic (e; von Kossa stain, original magnification ×40) and adipogenic differentiation (f; Oil Red, original magnification ×100)
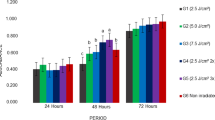
Analysis of the number of viable cells by the Trypan blue exclusion method revealed that the dose of 1.0 J/cm2 promoted an increase in cell proliferation at 48 and 72 h when compared to the control and Laser 0.5 groups (p < 0.05). Cell proliferation was lower in the control group compared to the irradiated groups (Laser 0.5 and Laser 1.0), with a statistically significant difference (p < 0.05) between the irradiated groups at 72 h (Fig. 3).

Number of SHEDs at the different time points studied. The same lowercase letters indicate a statistically significant difference (p < 0.05; Mann–Whitney test)
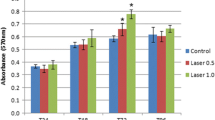
The results of mitochondrial activity measured by the MTT assay were similar to the Trypan blue counts. In this respect, a significantly greater reduction of MTT was observed in the group irradiated with the dose of 1.0 J/cm2 at 24, 48, and 72 h when compared to the non-irradiated group. A significant difference between the irradiated groups was only observed at 48 h (Table 1).
The distribution of cells across the different cell cycle phases was consistent with proliferating cells throughout the experiment, with a predominance of cells in the S and G2/M phases in the group irradiated with 1.0 J/cm2 at T48 and T72 (Fig. 4a–d). At 72 h after the first irradiation, expression of nuclear protein Ki67 was higher in cells of the irradiated groups compared to the control, with the dose of 1.0 J/cm2 promoting the highest expression of Ki67 (Fig. 4e). This result corroborates the findings of cell cycle analysis, showing that cells of the Laser 1.0 group were in the proliferative phase.

a–d Distribution of SHEDs across the phases of the cell cycle in the different groups at the time points analyzed. The percentage of each phase represents the mean of triplicate experiments. e Immunoexpression of Ki67 in the groups studied. FlowJo (USA)
Regarding the evaluation of apoptosis, the cells exhibited low levels of fluorescence for annexin V and PI by flow cytometry—which is compatible with viable cells—at 72 h, demonstrating the absence of significant alterations throughout the experiment (Fig. 5a–c). This finding indicates that the doses studied did not cause cell damage in SHEDs. Cells submitted to laser therapy exhibited no significant morphological nuclear damage such as nuclear fragmentation or pyknotic nuclei over the period studied (Fig. 5d–f).

a–c Immunostaining of SHEDs with annexin V/PI at 72 h. Annexin V negative/PI positive (Q1), annexin V/PI positive (Q2), annexin V positive/PI negative (Q3), and annexin V/PI negative (Q4). a Non-irradiated control group. b Laser 0.5 group. c Laser 1.0 group. d–f Photomicrographs of SHEDs stained with DAPI after 72 h of culture. d Control. e Laser 0.5. f Laser 1.0 (DAPI staining, ×400)
Discussion
Dental stem cells are isolated from dental alveolar tissues [17, 18] and are characterized by high proliferative capacity and plasticity. These cells can be used for the regeneration of dental facial structures (e.g., alveolar bone, dentin pulp complex) or other tissues such as neural tissue [19]. Human deciduous teeth have been reported as a promising source of mesenchymal stem cells, which can be used in different clinical applications such as dental tissue engineering [1], repair of bone defects [20], and even the treatment of neural tissue damage and degenerative diseases [21, 22].
The simple and convenient isolation of SHEDs and non-significant immunogenicity, which permits their use in allogenic transplants without the need for immunosuppressors [21, 23, 24], confer important clinical utility, including the creation of cell banks. However, the proliferation of these cells is a time-consuming process, and the development of methods that increase the number of available cells within a short period of time is fundamental. Within this context, one possible approach suggested in the literature is LLLI [25,26,27].
The biostimulating effect of LLLI has been reported for more than four decades. Since its first description, researchers in this field have tried to develop the best laser therapy protocol that would promote positive biostimulating effects attributed to low-level lasers [28]. Few of the various sources of stem cells have been submitted to laser irradiation [11,12,13], especially stem cells of dental origin. Thus, further studies using stems cells obtained from other sources are needed to better understand the effect of laser therapy on the proliferation of these cells. The present study evaluated the effect of LLLI on SHEDs, a source of dental stem cells that has been poorly studied, with only three published reports [28,29,30].
Regarding the effect of LLLI on pulp stem cells, positive effects of laser therapy on stem cells derived from the pulp of permanent teeth have been reported [27, 31, 32]. Investigation of the treatment of SHEDs with LLLI has shown positive effects on the survival of these cells under stress conditions [30] and on dental osteogenic differentiation [29]. However, this is the first study that investigated, in addition to cell viability and proliferation, the effects of laser therapy on cell cycle, apoptosis, and nuclear morphology, and favorable results regarding these biological events were obtained.
The choice of parameters of LLLI such as wavelength, power, and energy density influences the desirable proliferation results [26]. In this respect, both visible (red) and infrared lights exert biological effects on irradiated cells, and better results in terms of cellular biostimulation have been reported for the wavelength of 600 to 700 nm [33,34,35]. We therefore chose the wavelength of 660 nm, which promoted positive effects similar to those reported by Soares et al. [26], Zaccara et al. [27], and Eduardo et al. [31], confirming the biostimulating effect of the red light spectrum. In addition to the wavelength, the dose (energy density) is another parameter that needs to be considered when cellular biostimulation is desired. Doses ranging from 0.5 to 10 J/cm2 are used to induce cell proliferation, while doses higher than 10 J/cm2 exert antiproliferative effects [35]. Energy densities ranging from 0.5 to 4 J/cm2 have been more effective in stimulating the growth of stem cells [25,26,27, 36, 37], but the effect of varying energy density while maintaining the laser power constant is unknown for SHEDs.
According to Karu [38], an increase in energy density can damage photoreceptors, which reduces the biomodulatory effect of the laser. Lower doses therefore reduce the risk of cell damage and, in the case of stem cells, contribute to an increase in the population of these cells, maintaining their initial characteristics intact [26]. Using lower doses (0.5 and 1.0 J/cm2), laser therapy employed in the present study promoted biostimulation without causing damage to nuclear morphology or the loss of viability. This fact was confirmed by DAPI staining of the nuclei of irradiated cells, which demonstrated intact nuclei and the absence of nuclear fragmentation or pyknotic nuclei. In addition, analysis of apoptosis-related markers (annexin V/PI) showed that most of the cells subjected to laser therapy remained viable, supporting that LLLI does not affect the viability of the cells studied.
Our results corroborate the findings of Fernandes et al. [28] regarding the successful stimulation of proliferation of stem cells derived from the pulp of deciduous teeth using a wavelength of 660 nm and a low dose (1.2 J/cm2). However, the authors evaluated different combinations of dose and power (1.2 J/cm2–0.5 mW, 2.5 J/cm2–10 mW, 3.7 J/cm2–15 mW, 5.0 J/cm2–20 mW, and 6.2 J/cm2–25 mW), and cell proliferation was assessed after very short intervals (6 and 24 h). In fact, variations in the parameters of laser therapy have been one of the main obstacles to the comparison of studies on photobiomodulation [39].
Doses of 0.5 and 1.0 J/cm2 have been tested in studies on stem cells derived from the human periodontal ligament [26] and from the pulp of permanent teeth [27], with the other parameters of laser therapy being the same as those employed in the present study. Pulp stem cells from permanent teeth exhibited higher proliferation at the last time points studied (72 and 96 h after irradiation) when irradiated with the dose of 1.0 J/cm2 compared to the non-irradiated control group [27]. A similar behavior has been demonstrated for stem cells derived from human periodontal ligament, which exhibited a significantly higher proliferation rate when irradiated with a dose of 1.0 J/cm2 compared to the non-irradiated group at the last time points evaluated (48 and 72 h after irradiation) [26]. These results and the present findings suggest that the dose of 1.0 J/cm2 is effective in increasing the proliferation of other sources of stem cells. Furthermore, this dose resulted in a more marked proliferative response of SHEDs when compared to the dose of 0.5 J/cm2, as demonstrated by the expression of Ki67 and cell cycle distribution of cells.
Cell cycle analysis demonstrated the distribution of cells in the phases consistent with cell proliferation (S and G2/M), corroborating previous findings obtained for dental pulp stem cells [27]. Although the exact molecular mechanism whereby LLLI exerts its effects on cell proliferation is not fully understood, experimental data show that irradiation is followed by an increase in the synthesis of growth factors, nitric oxide, reactive oxygen species, ATP, RNA, and DNA [40]. In the MTT assay, the group irradiated with the dose of 1.0 J/cm2 exhibited better results at 24, 48, and 72 h after the first irradiation compared to the non-irradiated (control) group and performed significantly better than the group irradiated with the dose of 0.5 J/cm2 at 48 h. These results agree with Barboza et al. [25] who irradiated bone marrow and adipose tissue stem cells and Stein et al. [41] who irradiated human osteoblasts. Both studies reported increased cell proliferation when a wavelength of 660 nm and a dose of 1.0 J/cm2 were used.
Returning to the hypothesis posed at the beginning of this paper, it is now possible to state that LLLI under the conditions used in this study (power of 30 mW, wavelength of 660 nm, and energy density of 1.0 J/cm2) stimulates the proliferation of SHEDs without affecting cell viability. These data have potential clinical relevance since LLLI represents a therapeutic opportunity, especially in the field of cell therapy and tissue engineering. Considering the limited number of experiments and heterogeneity of the methods employed, further studies are needed to establish the ideal parameters of laser therapy for stimulating the differentiation of this type of cells.
References
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S (2003) SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A 100:5807–5812
Nakamura S, Yamada Y, Katagiri W, Sugito T, Ito K, Ueda M (2009) Stem cell proliferation pathways comparison between human exfoliated deciduous teeth and dental pulp stem cells by gene expression profile from promising dental pulp. J Endod 35:1536–1542
Arora V, Arora P, Munshi AK (2009) Banking stem cells from human exfoliated deciduous teeth (SHED): saving for the future. J Clin Pediatr Dent 33:289–394
Horvát-Karajz K, Balogh Z, Kovács V, Drrernat AH, Sréter L, Uher F (2009) In vitro effect of carboplatin, cytarabine, paclitaxel, vincristine, and low-power laser irradiation on murine mesenchymal stem cells. Lasers Surg Med 41:463–469
Wu Y, Wang J, Gong D, HY G, Hu S, Zhang H (2012) Effects of low-level laser irradiation on mesenchymal stem cell proliferation: a microarray analysis. Lasers Med Sci 27:509–519
Moore P, Ridgway TD, Higbee RG, Howard EW, Lucroy MD (2005) Effect of wavelength on low-intensity laser irradiation stimulated cell proliferation in vitro. Lasers Surg Med 36:8–12
Evans DH, Abrahamse H (2008) Efficacy of three laser wavelengths for in vitro wound healing. Photodermatol Photoimmunol Photomed 24:199–210
Schindl A, Merwald H, Schindl L, Kaun C, Wojta J (2003) Direct stimulatory effect of low-intensity 670 nm laser irradiation on human endothelial cell proliferation. Br J Dermatol 148:334–336
Fujihara NA, Hiraki KR, Marques MM (2006) Irradiation at 780 nm increases proliferation rate of osteoblasts independently of dexamethasone presence. Lasers Surg Med 38:332–336
AlGhamdi KM, Kumar A, Moussa NA (2012) Low-level laser therapy: a useful technique for enhancing the proliferation of various cultured cells. Lasers Med Sci 27:237–249
Ginani F, Soares DM, Barreto MP, Barboza CA (2015) Effect of low level laser therapy on mesenchymal stem cell proliferation: a systematic review. Lasers Med Sci 30:2189–2194
Marques MM, Diniz IM, de Cara SP, Pedroni AC, Abe GL, D’Almeida-Couto RS, Lima PL, Tedesco TK, Moreira MS (2016) Photobiomodulation of dental derived mesenchymal stem cells: a systematic review. Photomed Laser Surg 34:500–508
Borzabadi-Farahani A (2016) Effect of low-level laser irradiation on proliferation of human dental mesenchymal stem cells: a systemic review. J Photochem Photobiol B 162:577–582
Vasconcelos RG, Ribeiro RA, Vasconcelos MG, Lima KC, Barboza CA (2012) In vitro comparative analysis of cryopreservation of undifferentiated mesenchymal cells derived from human periodontal ligament. Cell Tissue Bank 13:461–469
Ginani F, Soares DM, Rabêlo LM, Rocha HAO, de Souza LB, Barboza CAG (2016) Effect of a cryopreservation protocol on the proliferation of stem cells from human exfoliated deciduous teeth. Acta Odontol Scand 74:598–604
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Dj P, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Cao Y, Song M, Kim E, Shon W, Chugal N, Bogen G, Lin L, Kim RH, Park NH, Kang MK (2015) Pulp-dentin regeneration: current state and future prospects. J Dent Res 94:1544–1551
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A 97:13625–13630
Liu J, Yu F, Sun Y, Jiang B, Zhang W, Yang J, GT X, Liang A, Liu S (2015) Concise reviews: characteristics and potential applications of human dental tissue-derived mesenchymal stem cells. Stem Cells 33:627–638
Zheng Y, Liu Y, Zhang CM, Zhang HY, Li WH, Shi S, Le AD, Wang SL (2009) Stem cells from deciduous tooth repair mandibular defect in swine. J Dent Res 88:249–254
Huang YY, Chen AC, Carroll JD, Hamblin MR (2009) Biphasic dose response in low-level light therapy. Dose Response 7:358–383
Morsczeck C, Völlner F, Saugspier M, Brandl C, Reichert TE, Driemel O, Schmalz G (2010) Comparison of human dental follicle cells (DFCs) and stem cells from human exfoliated deciduous teeth (SHED) after neural differentiation in vitro. Clin Oral Investig 14:433–440
Pierdomenico L, Bonsi L, Calvitti M, Rondelli D, Arpinati M, Chirumbolo G, Becchetti E, Marchionni C, Alviano F, Fossati V, Staffolani N, Franchina M, Grossi A, Bagnara GP (2005) Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation 80:836–842
Noël D, Djouad F, Bouffi C, Mrugala D, Jorgensen C (2007) Multipotent mesenchymal stromal cells and immune tolerance. Leuk Lymphoma 48:1283–1289
Barboza CA, Ginani F, Soares DM, Henriques AC, Freitas Rde A (2014) Low-level laser irradiation induces in vitro proliferation of mesenchymal stem cells. Einstein (Sao Paulo) 12:75–81
Soares DM, Ginani F, Henriques AG, Barboza CA (2015) Effects of laser therapy on the proliferation of human periodontal ligament stem cells. Lasers Med Sci 30:1171–1174
Zaccara IM, Ginani F, Mota-Filho HG, Henriques ÁC, Barboza CA (2015) Effect of low-level laser irradiation on proliferation and viability of human dental pulp stem cells. Lasers Med Sci 30:2259–2264
Fernandes AP, Junqueira Mde A, Marques NC, Machado MA, Santos CF, Oliveira TM, Sakai VT (2016) Effects of low-level laser therapy on stem cells from human exfoliated deciduous teeth. J Appl Oral Sci 24:332–337
Turrioni AP, Basso FG, Montoro LA, Almeida Lde F, Costa CA, Hebling J (2014) Phototherapy up-regulates dentin matrix proteins expression and synthesis by stem cells from human-exfoliated deciduous teeth. J Dent 42:1292–1299
Diniz IM, Matos AB, Marques MM (2015) Laser phototherapy enhances mesenchymal stem cells survival in response to the dental adhesives. Sci World J 2015:671789
Eduardo Fde P, Bueno DF, de Freitas PM, Marques MM, Passos-Bueno MR, Eduardo Cde P, Zatz M (2008) Stem cell proliferation under low-intensity laser irradiation: a preliminary study. Lasers Surg Med 40:433–438
Pereira LO, Longo JPF, Azevedo RB (2012) Laser irradiation did not increase the proliferation or the differentiation of stem cells from normal and inflamed dental pulp. Arch Oral Biol 57:1079–1085
Tuby H, Maltz L, Oron U (2007) Low-level laser irradiation (LLLI) promotes proliferation of mesenchymal and cardiac stem cells in culture. Lasers Surg Med 39:373–378
Soleimani M, Abbasnia E, Fathi M, Sahraei H, Fathi Y, Kaka G (2012) The effects of low-level laser irradiation on differentiation and proliferation of human bone marrow mesenchymal stem cells into neurons and osteoblasts: an in vitro study. Lasers Med Sci 27:423–430
AlGhamdi KM, Kumar A, Mouss NA (2012) Low level laser therapy: a useful technique for enhancing the proliferation of various cultured cells. Lasers Med Sci 27:237–249
Mvula B, Mathope T, Moore T, Abrahamse H (2008) The effect of low-level laser irradiation on adult human adipose-derived stem cells. Lasers Med Sci 23:277–282
Mvula B, Moore TJ, Abrahamse H (2010) Effect of low-level laser irradiation and epidermal growth factor on adult human adipose-derived stem cells. Lasers Med Sci 25:33–39
Karu TI (1987) Photobiological fundamentals of low-power laser therapy. J Quantum Electron 23:1703–1717
Tunér J, Jenkins PA (2016) Parameter reproducibility in photobiomodulation. Photomed Laser Surg 34:91–92
Gao X, Xing D (2009) Molecular mechanisms of cell proliferation induced by low power laser irradiation. J Biomed Sci 16:4
Stein A, Benayahu D, Maltz L, Oron U (2005) Low-level laser irradiation promotes proliferation and differentiation of human osteoblasts in vitro. Photomed Laser Surg 23:161–166
Funding
This study was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Grant No. 480958/2012-2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in the study were in accordance with the Ethics Committee of the Federal University of Rio Grande do Norte (Approval No. 34913314.4.0000.5537) and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Ginani, F., Soares, D.M., de Oliveira Rocha, H.A. et al. Low-level laser irradiation induces in vitro proliferation of stem cells from human exfoliated deciduous teeth. Lasers Med Sci 33, 95–102 (2018). https://doi.org/10.1007/s10103-017-2355-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-017-2355-y