
Abstract
A positive effect of low-level laser irradiation (LLLI) on the proliferation of some cell types has been observed, but little is known about its effect on dental pulp stem cells (DPSCs). The aim of this study was to identify the lowest energy density able to promote the proliferation of DPSCs and to maintain cell viability. Human DPSCs were isolated from two healthy third molars. In the third passage, the cells were irradiated or not (control) with an InGaAlP diode laser at 0 and 48 h using two different energy densities (0.5 and 1.0 J/cm²). Cell proliferation and viability and mitochondrial activity were evaluated at intervals of 24, 48, 72, and 96 h after the first laser application. Apoptosis- and cell cycle-related events were analyzed by flow cytometry. The group irradiated with an energy density of 1.0 J/cm² exhibited an increase of cell proliferation, with a statistically significant difference (p < 0.05) compared to the control group at 72 and 96 h. No significant changes in cell viability were observed throughout the experiment. The distribution of cells in the cell cycle phases was consistent with proliferating cells in all three groups. We concluded that LLLI, particularly a dose of 1.0 J/cm², contributed to the growth of DPSCs and maintenance of its viability. This fact indicates this therapy to be an important future tool for tissue engineering and regenerative medicine involving stem cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells have the capacity of self-renewal and of developing into tissues compatible with their origin. These characteristics are recognized as important functions [1]. These cells have been isolated from oral tissues such as dental pulp [2–4], dental pulp of deciduous teeth [5, 6], and periodontal ligament [2, 7, 8]. Considered to be a relatively rich source of mesenchymal stem cells, interest in the isolation and banking of dental pulp stem cells (DPSCs) has increased substantially in recent years [9]. These cells represent a source of therapeutic cells that permit a more physiological regeneration of the dentin-pulp complex and may also be used for tissue engineering of a whole “biotooth” [10].
Low-level laser irradiation (LLLI) promotes the biomodulation and proliferation of a variety of cell types [11, 12]. Previous studies have shown that LLLI increases the proliferation rates of bone marrow [13], cardiac tissue [11], adipose tissue [14], and periodontal ligament cells [15]. However, very little is known about the biological activity of DPSCs submitted to LLLI.
Alghamdi et al. [16] have shown that the application of LLLI at an energy density of 0.5 to 4.0 J/cm2 and visible spectrum of 600 to 700 nm increases the proliferation rate of various cell lines. According to Karu [17], a higher energy density can damage photoreceptors, with a consequent reduction in the biomodulatory effect of the laser. Therefore, the objective of the present study was to identify the lowest energy density able to promote the proliferation of DPSCs. We also hypothesized that LLLI can improve the effectiveness of DPSC proliferation and maintain cell viability.
Materials and methods
Subjects
The study was approved by the Ethics Committee of the Federal University of Rio Grande do Norte, Brazil. Human DPSCs were obtained from two healthy permanent third molars extracted due to a surgical/orthodontic indication. The teeth were obtained from patients who presented good systemic and oral health. After extraction, the teeth were abraded at the enamel/cementum junction with a diamond bur (#2134, KG Sorensen, Brazil), avoiding direct contact of the bur with the pulp. Next, the crown and the root were separated with orthodontic pliers in order to expose the pulp to the culture medium.
Cell culture and irradiation
Crowns and roots were stored in 50-mL Falcon tubes (TPP®, Switzerland) containing 5 mL α-MEM (Life Technologies, USA) under hypothermic conditions (4 °C) for up to 2 h, until the processing of pulp under sterile conditions. Next, the crowns and the roots were washed three times (10 min each) in a solution containing α-MEM supplemented with 10,000 IU/mL penicillin, 10,000 μg/mL streptomycin, 100 mg/mL gentamicin, and 250 μg/mL amphotericin B (all antibiotics were from Life Technologies) to eliminate possible contamination.
The dental pulp was carefully removed using a Hedstroem file (Dentsply, Brazil) and then submitted to enzymatic digestion with 3 mg/mL Collagenase I (Life Technologies) and 4 mg/mL Dispase (Life Technologies, USA) for 1 h at 37 °C. The cells were cultured on plates containing α-MEM supplemented with 15 % fetal bovine serum (Life Technologies). The cultures were maintained at 37 °C in a 5 % CO2 atmosphere, and the culture medium was changed at intervals of 3 days until the cells reached 70–95 % confluence.
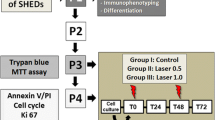
In the second passage (P2), the cells were analyzed to confirm their stem cell nature, according to the criteria established by Dominici et al. [18]. Briefly, an aliquot of cells was evaluated by flow cytometry using the Human MSC Analysis Kit (BD Stemflow™, USA), which revealed positive staining for surface markers of mesenchymal stem cells (CD105 PerCP-Cy5.5, CD73-APC, CD90-FITC) and negative staining for markers of hematopoietic stem cells (CD45, CD34, CD11b, CD19, HLA-DR). Additionally, the multilineage differentiation potential of DPSCs was confirmed by culturing the cells in osteogenic and adipogenic differentiation media (StemPro® Differentiation Kits, Invitrogen, USA) for up to 21 days. Light microscopy analysis showed the deposition of mineralized matrix stained by von Kossa and lipid vesicles stained by Oil Red O, what represents, respectively, the differentiation of osteoblastic and adipose cells.
In the third passage (P3), DPSCs were divided into three groups according to treatment: (i) control group: no irradiation; (ii) laser 0.5: cells irradiated with an energy density of 0.5 J/cm2; and (iii) laser 1.0: cells irradiated with an energy density of 1.0 J/cm2. An InGaAlP diode laser (Kondortech, Brazil) operating at the following parameters was used: power of 30 mW, wavelength of 660 nm, continuous action mode, and a tip diameter of 0.01 cm2. The cells were irradiated at 0 and 48 h. The laser probe was fixed perpendicular to each plate at a distance of 0.5 cm from the cells. The cells were plated in such a way that there was always one (for 24-well plates) or three (for 96-well plates) empty wells between seeded wells in order to minimize the unintentional dispersion of light between wells during laser application.
Analysis of cell proliferation
Cell viability and proliferation were analyzed at 24, 48, 72, and 96 h after the first laser application in the control (not irradiated) and irradiated groups by the MTT assay (evaluation of mitochondrial activity by reduction of MTT) and by the Trypan blue exclusion method. For the MTT assay, the cells were cultured in 96-well plates at a density of 5 × 103 cells/well. One 96-well plate was used for each time point, with four wells per group (control, 0.5 and 1.0 J/cm2), and absorbance of the samples was monitored in an ELISA reader at 570 nm. For Trypan blue staining, the cells were cultured in 24-well plates at a density of 3 × 104 cells/well. The number of Trypan blue-stained cells in each well was counted in a Neubauer chamber at each time point. The cell counts were performed by a blinded and previously calibrated examiner.
Analysis of apoptosis and cell cycle
Apoptosis was evaluated by flow cytometry (Invitrogen, USA) using the FITC Annexin V/Dead Cell Apoptosis Kit with FITC annexin and propidium iodine (PI). For this purpose, the cells were cultured in 6-well plates at a density of 2 × 105 cells/well. After 24 and 72 h (corresponding to the first 24 h after each application), the cells were washed in cold phosphate-buffered saline (PBS) and resuspended in 200 μL binding buffer (1×). The cells were stained with 3 μL annexin V-FITC and 1 μL PI (100 μg/mL) and incubated for 15 min. Next, 400 μL binding buffer for annexin V (1×) was added, and the cells were analyzed by flow cytometry.
For cell cycle analysis, an aliquot of the cells was incubated with 2 % paraformaldehyde, washed in cold PBS, and permeabilized with 0.01 % saponin for 15 min. After this procedure, the cells were incubated with 10 μL RNAse (4 mg/mL) for 40 min at 37 °C. Next, 5 μL PI (25 mg/mL) and 200 μL cold PBS were added, and the cells were analyzed by flow cytometry.
Statistical analysis
Differences between groups at each time point studied were analyzed by the Kruskal-Wallis and Mann-Whitney tests, considering a level of significance of 5 % (p < 0.05).
Results
Effect of LLLI on cell proliferation
The number of DPSCs analyzed by the Trypan blue exclusion method in the different groups is shown in Table 1. An increase in cell proliferation over time was observed in all groups. Analysis of the mean number of cells showed a higher proliferation rate in the irradiated groups when compared to the control group, with a statistically significant difference (p < 0.05) at 72 h. At 96 h, a significant difference between groups was only observed for the group irradiated with an energy density of 1.0 J/cm2. No difference in cell viability analyzed by the Trypan blue exclusion method was observed between groups at any of the time points studied.
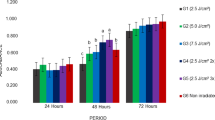
The pattern of mitochondrial activity of DPSCs analyzed by the MTT assay is illustrated in Fig. 1. In the irradiated groups, mitochondrial activity followed the same trend as observed by the Trypan blue exclusion method. Irradiation with energy densities of 0.5 and 1.0 J/cm2 resulted in a significantly larger number of cells when compared to the control group after the second irradiation at an interval of 72 h.

Proliferation of human dental pulp stem cells incubated for different periods of time evaluated by MTT assay. Values are the mean percentage ± standard deviation (*p < 0.05, Mann-Whitney test)
Effect of LLLI on apoptosis
The dot plots obtained by flow cytometry analysis are shown in Fig. 2. The cells exhibited weak positive staining for annexin V and PI, both markers of apoptosis. No significant changes in cell viability were observed throughout the experiment, although there was a slight increase in percent viability in all groups after the second application of LLLI.

Immunostaining of dental pulp stem cells with annexin V/propidium iodide (PI) at intervals of 24 h (a control, b laser 0.5 J/cm2, and c laser 1.0 J/cm2) and 72 h (d control, e laser 0.5 J/cm2, and f laser 1.0 J/cm2)
Effect of LLLI on the cell cycle
The distribution of cells in the cell cycle phases is shown in Fig. 3, which illustrates the percentage of cells in each phase of the cycle. At 24 h after plating, there was a higher percentage of cells in G0/G1 (more than 50 %), but no statistically significant difference (p > 0.05) was observed between groups. At the last time point examined, approximately 85 % of the cells were in the S and G2/M phases, with no significant differences between groups, a finding that was consistent with proliferating cells in all three groups.

Distribution of dental pulp stem cells in the cell cycle phases (G0/G1, S, and G2/M) for the three groups over time
Discussion
The role of stem cells in tissue repair has been extensively studied due to their self-renewal and differentiation properties [11]. DPSCs have been shown to maintain their characteristics after cryopreservation, to differentiate into bone-like tissues when loaded on scaffolds in animal models, and to regenerate bone in human grafts [19]. These cells are also able to differentiate into functional odontoblasts and vascular endothelial cells, suggesting that they can serve as a single source for dental pulp tissue engineering [9]. However, one requisite for the use of stem cells in regenerative medicine is their availability in abundant quantities. Within this context, LLLI has been shown to induce stem cell activity by increasing cell migration, proliferation, and viability, activating protein expression, and inducing the differentiation of progenitor cells [20].
Some aspects of the laser can influence the desired proliferation results, such as the ideal light spectrum, power level, energy density, and wavelength [15]. Peplow et al. [21] argue that comparison between the results of previous studies is difficult due to the wide range of irradiation parameters, methodologies, and cell types used. Recent studies have shown that the best results are obtained when the visible light spectrum (600 to 700 nm) is used [11]. In contrast, the infrared light spectrum (810 to 830 nm) has been associated with the inhibition of proliferation [22]. We chose a wavelength of 660 nm, which promoted positive stimulatory effects similar to those reported by Soares et al. [15], Eduardo et al. [23], and Horvát-Karajz et al. [24].
The effect of LLLI on DPSCs has been investigated by Eduardo et al. [23], who noted that an energy density of 3.0 J/cm2 associated with a wavelength of 660 nm positively stimulated cell proliferation. Using a lower energy density (1.0 J/cm2), Soares et al. [15] observed a biostimulatory effect of LLLI on periodontal ligament stem cells. The energy density is an important factor that influences the results of cell proliferation. According to Karu [17], an increase in energy density may damage the photoreceptors, with a consequent reduction in the biomodulatory effect of LLLI. On the basis of these findings, we used the same parameters as Soares et al. [15] for the irradiation of DPSCs, including an InGaAlP laser, power of 30 mW, wavelength of 660 nm, and energy densities of 0.5 and 1.0 J/cm2, which promoted the biostimulation of DPSCs.
The number and interval between laser applications also influence the results of LLLI. Li et al. [25] compared the proliferation of bone marrow stem cells irradiated at intervals of 48 h over a period of 13 days and observed a positive effect on cell proliferation. Similar results have been reported by Soares et al. [15] who used a power of 30 mW and energy density of 1.0 J/cm2. A lower energy density of 0.5 J/cm2 showed little influence. In the present study, the response was more pronounced when the cells were irradiated with the dose of 1.0 J/cm2 compared to 0.5 J/cm2, thus confirming that, if an insufficient energy density is applied, no cellular response occurs because the threshold has not been reached. For the occurrence of biostimulation, the threshold should be achieved by applying more energy [26].
Laser therapy exerts a dose-dependent effect on biological responses and seems to have a cumulative effect at each new dose applied [26]. This fact was confirmed in the present study by observation of the increase of cell proliferation rates at the last two time points (72 and 96 h) after the second irradiation, what represents a cumulative effect over time at the dose of 1.0 J/cm2.
Hawkins and Abrahamse [27] submitted fibroblasts to LLLI and observed increased cell viability beyond acceleration of the proliferation process. With respect to viability, similar results were found in the present study. Indeed, cell counts and analysis of apoptosis-related markers (annexin V/PI) showed that most of the cells remained viable and percent viability even increased in all groups after the second application of LLLI.
The decrease in the percentage of cells in G0/G1 associated with an increase in the percentage of cells in the S and G2/M phases indicates the transfer from G1 to S and G2/M and, consequently, cell proliferation [28]. In the present study, cell cycle analysis confirmed the results of the other tests, with the distribution of cells in the cell cycle phases being consistent with cell proliferation. Although the exact molecular mechanism by which LLLI exerts its effects on cell proliferation is not completely understood, experimental data showed that irradiation is followed by increased synthesis of growth factors, nitric oxide (NO), reactive oxygen species (ROS), ATP, RNA, and DNA [29].
The results of this study have potential clinical relevance since the use of LLLI represents a therapeutic opportunity in dentistry, especially in the field of dental pulp tissue engineering associated with stem cells. In light of future cell therapy protocols, laser therapy permits to significantly increase the initial number of stem cells before differentiation, thus increasing the number of differentiated cells for tissue engineering and regenerative and healing processes.
In summary, the present study showed that the parameters of LLLI (30 mW power, 660 nm wavelength, and 1.0 J/cm2 energy density) promoted cell proliferation. Laser irradiation did not affect cell viability throughout the experiment.
References
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A 97:13625–13630
Gronthos S, Mrozik K, Shi S, Bartold PM (2006) Ovine periodontal ligament stem cells: isolation, characterization, and differentiation potential. Calcif Tissue Int 79:310–317
Batouli S, Miura M, Brahim J, Tsutsui TW, Fisher LW, Gronthos S, Robey PG, Shi S (2003) Comparison of stem-cell-mediated osteogenesis and dentinogenesis. J Dent Res 82:976–981
Sloan AJ, Smith AJ (2007) Stem cells and the dental pulp: potential roles in dentine regeneration and repair. Oral Dis 13:151–157
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S (2003) SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A 100:5807–5812
Kerkis I, Kerkis A, Dozortsev D, Stukart-Parsons GC, Gomes Massironi SM, Pereira LV, Caplan AI, Cerruti HF (2006) Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells Tissues Organs 184:105–116
Seo BM, Miura M, Gronthos S, Bartold PM, Batouli S, Brahim J, Young M, Robey PG, Shi S (2004) Investigation of multipotent stem cells from human periodontal ligament. Lancet 364:149–155
Chen SC, Marino V, Gronthos S, Bartold PM (2006) Location of putative stem cells in human periodontal ligament. J Periodontal Res 41:547–553
Piva E, Silva AF, Nör JE (2014) Functionalized scaffolds to control dental pulp stem cell fate. J Endod 40:S33–S40. doi:10.1016/j.joen.2014.01.013
Schmalz G, Smith AJ (2014) Pulp development, repair, and regeneration: challenges of the transition from traditional dentistry to biologically based therapies. J Endod 40:S2–S5. doi:10.1016/j.joen.2014.01.018
Tuby H, Maltz L, Oron U (2007) Low-level laser irradiation promotes proliferation of mesenchymal and cardiac stem cells in culture. Lasers Surg Med 39:373–378
Wu Y, Wang J, Gong D, Gu H, Hu S, Zhang H (2012) Effects of low-level laser irradiation on mesenchymal stem cell proliferation: a microarray analysis. Laser Med Sci 27(2):509–519. doi:10.1007/s10103-011-0995-x
Hou JF, Zhang H, Yuan X, Li J, Wei YJ, Hu SS (2008) In vitro effects of low-level laser irradiation for bone marrow mesenchymal stem cells: proliferation, growth factors secretion and myogenic differentiation. Lasers Surg Med 40(10):726–733. doi:10.1002/lsm.20709
Mvula B, Mathope T, Moore T, Abrahamse H (2008) The effect of low-level laser irradiation on adult human adipose-derived stem cells. Lasers Med Sci 23(3):277–282
Soares DM, Ginani F, Henriques AG, Barboza CA (2013) Effects of laser therapy on the proliferation of human periodontal ligament stem cells. Lasers Med Sci doi: 10.1007/s10103-013-1436-9.
Alghamdi KM, Kumar A, Mouss NA (2012) Low-level laser therapy: a useful technique for enhancing the proliferation of various cultured cells. Lasers Med Sci 27:237–249. doi:10.1007/s10103-011-0885-2
Karu TI (1987) Photobiological fundamentals of low-power laser therapy. J Quantum Electron 23:1703–1817
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
La Noce M, Paino F, Spina A, Naddeo P, Montella R, Desiderio V, De Rosa A, Papaccio G, Tirino V, Laino L (2014) Dental pulp stem cells: state of the art and suggestions for a true translation of research into therapy. J Dent 42:761–768. doi:10.1016/j.jdent.2014.02.018
De Villiers JA, Houreld NN, Abrahamse H (2011) Influence of low intensity laser irradiation on isolated human adipose derived stem cells over 72 hours and their differentiation potential into smooth muscle cells using retinoic acid. Stem Cell Rev 7(4):869–882. doi:10.1007/s12015-011-9244-8
Peplow PV, Chung TY, Baxter GD (2010) Laser photobiomodulation of proliferation of cells in culture: a review of human and animal studies. Photomed Laser Surg 28:S3–S40. doi:10.1089/pho.2010.2771
Hawkins DH, Abrahamse H (2007) Time-dependent responses of wounded human skin fibroblasts following phototherapy. J Photochem Photobiol B 88:147–155
Eduardo FP, Bueno DF, de Freitas PM, Marques MM, Passos-Bueno MR, Eduardo CP, Zatz M (2008) Stem cell proliferation under low-intensity laser irradiation: a preliminary study. Lasers Surg Med. doi: 10.1002/lsm.20646.
Horvát-Karajz K, Balogh Z, Kovács V, Hámori Drrernat A, Sréter L, Uher F (2009) In vitro effect of carboplatin, cytarabine, paclitaxel, vincristine, and low-power laser irradiation on murine mesenchymal stem cells. Lasers Surg Med 41(6):463–469. doi:10.1002/lsm.20791
Li WT, Leu YC, Wu JL (2010) Red-light light-emitting diode irradiation increases the proliferation and osteogenic differentiation of rat bone marrow mesenchymal stem cells. Photomed Laser Surg. doi: 10.1089/pho.2009.2540.
Huang YY, Chen ACH, Carroll JD, Hamblin MR (2009) Biphasic dose response in low-level light therapy. Dose Response 7:358–383
Hawkins D, Abrahamse H (2006) Effect of multiple exposures of low-level laser therapy on the cellular responses of wounded human skin fibroblasts. Photomed Laser Surg 24:705–714
Schartinger VH, Galvan O, Riechelmann H, Dudás J (2012) Differential responses of fibroblasts, non-neoplastic epithelial cells, and oral carcinoma cells to low-level laser therapy. Support Care Cancer 20:523–529. doi:10.1007/s00520-011-1113-0
Gao X, Xing D (2009) Molecular mechanisms of cell proliferation induced by low power laser irradiation. J Biomed Sci 16:4. doi:10.1186/1423-0127-16-4
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Zaccara, I.M., Ginani, F., Mota-Filho, H.G. et al. Effect of low-level laser irradiation on proliferation and viability of human dental pulp stem cells. Lasers Med Sci 30, 2259–2264 (2015). https://doi.org/10.1007/s10103-015-1803-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-015-1803-9