
Abstract
Mixed connective tissue disease (MCTD) is a chronic autoimmune disease, which has a broad range of clinical manifestations shared by systemic lupus erythematosus, systemic sclerosis, polymyositis/dermatomyositis, and rheumatoid arthritis. MCTD is featured with high serum titers of anti-ribonucleoprotein antibodies and multiple system involvement. Its spinal cord involvement mainly manifests as transverse myelopathy (TM) and longitudinal extensive transverse myelopathy (LETM). Myelopathy in MCTD is extremely rare, and is usually characterized by serious neurological complications, such as paralysis or muscular paresis, sensory impairment, and smooth muscle dysfunction. Progressive clinical manifestations combined with laboratory examinations and magnetic resonance imaging examinations play important roles in the diagnosis of this disease. In order to prevent permanent neurological damage to the spinal cord, plasmapheresis and intravenous immunoglobulin can be performed in patients at the early disease stage. Early high-dose corticosteroids combined with cyclophosphamide, followed by low doses of immunosuppressors, can improve the long-term prognosis of patients. There are only nine global cases reported on MCTD associated with myelopathy at present. The death rate and disability rate of myelopathy in MCTD are extremely high. In this review, the pathomechanisms, clinical manifestations, auxiliary examination, diagnosis, differential diagnosis, treatment, and prognosis of myelopathy in MCTD were systematically elucidated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mixed connective tissue disease (MCTD) was first described in 1972 by Sharp et al. as a rare autoimmune disorder with multiple systems involvement [1]. Sharing some common clinical features with systemic lupus erythematosus (SLE) and polymyositis (PM), MCTD has been defined as a distinct disease [2, 3]. The involvement of the nervous system infrequently occurs in MCTD. Although the exact number remains unknown, the neurological disorder complicates the MCTD course in approximately 10% of patients [4]. The neurological manifestations of MCTD are usually mild. However, some patients with MCTD might have concurrent acute or subacute paraplegia, sensory deficits, dysfunction of the rectum and bladder sphincter, abnormal knee-jerk or tendon reflex, and other nervous system symptoms, which suggests the involvement of the spinal cord. Myelopathy can be diagnosed in these MCTD patients after excluding other diseases, such as infection, trauma, and tumor [5]. As an extremely rare complication of MCTD, myelopathy has been sporadically reported to be complicated with MCTD, and merely nine cases have been reported in the world at present. Due to the high rate of mortality and disability in MCTD complicated with myelopathy, wide attention has been given to this distinct clinical syndrome. In this review, the pathogenesis, clinical manifestations, laboratory and imaging examination, diagnosis, treatment, and prognosis of myelopathy in MCTD were systematically elaborated.
Mixed connective tissue disease
MCTD was first described in 1972 as a clinical syndrome with mixed features of SLE, polymyositis/dermatomyositis (PM/DM), systemic sclerosis (SSc), and rheumatoid arthritis (RA) and was identified to display high serum titers of anti-U1 ribonucleoprotein (anti-U1RNP) antibodies [1, 6].
MCTD has been considered to be the least common CTD, which is supported by a recent population-based epidemiology study conducted in Norway [7]. This study estimated a mean annual incidence of 2.1 per million per year and a point prevalence of adult-onset MCTD of 3.8 per 100,000 [7, 8]. The female-to-male ratio was 3.3 [7]. The observed female-to-male ratio in a large Hungarian cohort study reported a ratio of 12.3 [9].
Over the last years, significant advances have been made in disease pathogenesis understanding, and a central pathogenetic role of anti-U1RNP autoantibodies has clearly emerged [10,11,12]. It is believed that MCTD is mediated by the autoimmune response under the stimulation of external environmental factors on the basis of individual genetic background [13, 14]. Physiological stress factors, such as virus infection or ultraviolet radiation, lead to the release of a large number of apoptotic substances–containing RNP. In individuals with defects in eliminating these apoptotic particles, these particles encounter circulating immunoglobulin and dendritic cells; then after a series of immune processes, B lymphocytes are activated to secrete autoantibodies. These autoantibodies can recognize intact or modified individual U1 RNP proteins as well as structures composed of multiple subunits of the U1 RNP macromolecule and directly provoke the anti-RNP responses that lead to a series of related clinical manifestations [11, 15].
MCTD has a clinical spectrum including Raynaud’s phenomenon, swelling hands with sausage-like fingers, sclerodactyly, interstitial lung disease, esophagus dysmotility, and myositis [1, 16]. In addition, lymphadenopathy, malar rash, kidney damage, or alopecia are less common [6]. Other common and unspecific constitutional symptoms include myalgias, arthralgias, fatigue, and fever [17]. However, the above clinical manifestations are not unique to MCTD and can also occur in SLE, SSc, RA, and PM.
The diagnosis of MCTD is often not easy, because MCTD has a broad range of symptoms and signs. MCTD may be initiated with any clinical manifestations of SLE, SSc, PM, or RA during the disease progression. Clinical manifestations or laboratory alterations usually occur sequentially. Therefore, many patients do not meet the classification criteria for MCTD at the beginning of the disease, or only meet the classification criteria for other autoimmune diseases, such as SLE, SD, PDM, or RA.
In view of the heterogeneity of clinical manifestations, several MCTD classification proposals have been made [18,19,20]. At present, there are four coexisting diagnostic criteria for MCTD, including the criteria of Alarcon-Segovia and Villarreal, Kahn and Appelboom, Sharp, and Kasukawa, which are distinctly different and make the diagnosis of MCTD more difficult. In a recent study, Capelli S. et al. compared three different classification criteria for the diagnosis of MCTD (Kasukawa, Alarcón-Segovia, and Sharp) with defined predictors, including clinical features and autoantibody levels, which are potential shared by other CTDs [20, 21]. Their results revealed that the criteria from Kasukawa were more sensitive (75%), when compared with those from Alarcón-Segovia (73%) and Sharp (42%) [21]. To date, the concordance of clinical findings, serologic features, and alterations from close clinical follow-up is the strategy to approach a more accurate diagnosis of MCTD.
At present, although most investigators have considered MCTD as a distinct disease, some researchers still consider that MCTD belongs to subgroups of certain definite connective tissue disease (CTDs) or a transient phase before developing into another CTD, or in fact, overlap syndromes [3]. However, recent genetic studies have confirmed that MCTD is an evident human leukocyte antigen (HLA)-linked disease [10, 22]. The HLA profiles of MCTD patients distinctly differ from patients with other forms of CTDs. Specifically, MCTD is predominantly associated with HLA-DR4 (especially the loci DRB1*15:01, DRB1*04, and *09:01), whereas SLE is fundamentally correlated to HLA-DR2 and DR3 [22]. As SSc shows an association with HLA-DR3 or DR5, PM/DM also shows an association with HLA-DR3. Therefore, these genetic studies support the notion that MCTD is a distinct entity independent of other identified CTDs, such as SLE and SSc.
Myelopathy in MCTD
Neuropsychiatric (NP) involvement in patients with MCTD is a severe manifestation of the disease, which drastically impacts the quality of life of patients. Due to the high mortality and disability rate, NP symptoms have been considered as one of the major manifestations of the proposed disease activity criteria for MCTD [23]. Among patients with MCTD, the prevalence of NP symptoms varied from 5 to 39% [24,25,26]. The most frequently reported neurological manifestations are trigeminal neuropathy [27,28,29], headaches [12, 30, 31], aseptic meningitis [24, 27, 32, 33], cerebrovascular disease [27, 34, 35], and peripheral neuropathy [24, 27, 28, 36, 37], while sensory-neural hearing loss [38], depression [39, 40], cognitive impairment [41], psychosis [39], optic neuritis [25, 42, 43], and transverse myelopathy [1, 4, 43,44,45,46,47,48,49] have been less frequently reported. Furthermore, merely individual reports were distributed on restless leg syndrome [50], organic brain syndrome [51, 52], and posterior reversible encephalopathy syndrome [53].
Myelopathy is a rare and severe neurological complication of MCTD, in which merely nine cases have been reported at present (Table 1). MCTD complicated by myelopathy is mostly involved in young and middle-aged women, in which seven female patients in nine cases have been reported [4, 43,44,45,46,47, 49]. The average age of onset was 38.6 years old. Myelopathy can occur in the early stage of MCTD, since six cases (66.7%) developed myelopathy within 2 years from the onset of MCTD [4, 44, 46,47,48,49]. However, myelopathy was not identified as the first symptom in these patients with MCTD. The course of myelopathy in MCTD can be manifested as progressive aggravation, which progresses to the disease peak within several days, and sometimes even months.
Myelopathy in MCTD may present as transverse myelopathy (TM) with the involvement of less than 3 segments of the spinal cord, or as longitudinal extensive transverse myelopathy (LETM) [54], in which more than three segments (continuous or not) are involved. The main clinical manifestations include limb paralysis, muscle paralysis, sensory deficits, and sphincter-dominated smooth muscle dysfunction. In recent years, with the advancements in imaging technology, LETM was noted in one case of MCTD complicated by myelopathy through magnetic resonance imaging (MRI) examination.
Retrospective analysis revealed that all nine reported patients with myelopathy had inflammatory lesions that involved the thoracic spinal cord, and merely one case had lesions that involved both the cervical and thoracic spinal cord. This result suggests that the thoracic region is most frequently affected. Moreover, myelopathy was accompanied by other neurological manifestations in MCTD patients. For example, optic neuritis has been reported in two of nine previously reported patients [4], and depression and peripheral neuropathy have been reported in one of them [47, 48].
Myelopathy is one of 19 NP manifestations in SLE [55]. Compared with myelopathy in MCTD, more cases of myelopathy have been reported in SLE patients (Table 2). The incidence of myelopathy in SLE patients is 1–2% [55, 61], and most of these incidents occurred within 5 years after the onset of SLE [62], approximately 50% of SLE patients manifested myelopathy as the first symptom [56]. Apart from the thoracic region, almost 50% of patients with TM may have inflammatory lesions involved in the cervical spinal cord [56]. The course of myelopathy in SLE can manifest as acute aggravation, and develop to a peak in several hours or days. Complete or partial injury of the spinal cord can occur within 24 h, which results in a dramatically acute course [56,57,58]. LETM is more frequently reported in patients with SLE [59, 63]. In addition, more recurrences were found in SLE patients without treatments, or those treated with long-term, low or medium doses of glucocorticosteroids [64]. The episodes of at least one recurrence were noted in 21–55% of patients with myelopathy in SLE [56, 64, 65].
Taken together, compared with myelopathy in SLE, myelopathy in MCTD is relatively mild and progressively aggravated. The extent of inflammatory injury in MCTD is limited mainly to the thoracic spinal cord, and the recurrence rate is low, which may suggest that the prognosis of the myelopathy in MCTD is better than that in SLE.
Pathomechanism
As an inflammation-associated chronic disease, MCTD can be distinguished through the production of pathogenic autoantibodies that are resulted from perturbed immune responses in the host. At present, the specific pathomechanism of myelopathy in MCTD remains unclear. It is possible that multiple mechanisms are involved, including genetic factors, blood brain barrier dysfunction, vasculitis, vascular occlusion, autoantibody-mediated tissue and neuroendocrine-immune imbalance, neuronal damage, inflammatory mediators, and direct neuronal cell death, which makes the pathogenesis of myelopathy in MCTD very complex.
The autopsy results of a MCTD patient with myelopathy revealed focal infarction of the spinal cord parenchyma, glial cell proliferation, and thickening of the vascular wall, with a large amount of infiltrated inflammatory cells dominated by plasma cells [4]. The disintegration of the nerve axon and demyelination of the spinal cord were observed under a microscope [4]. Circulating anti-phospholipid antibody was also observed. Therefore, it has been postulated that the vasculitis of small arachnoid arteries of the spinal cord and arterial thrombosis may cause spinal cord micro-infarction. The involvement of the thoracic spinal cord may be due to its poor vascularization and pathological changes, such as ischemia and necrosis, would more likely occur when vasculitis happens.
Accumulating evidence has indicated that MCTD patients may have higher titers of anti-endothelial cell antibodies (AECA) and antibodies against phospholipid structures, such as cardiolipin (anti-CL) and β2-glycoprotein I (anti-β2GPI) [66, 67]. MCTD patients complicated with CVD were found to have increased titers of anti-U1RNP, IgG and IgM anti-CL, and AECA [67]. Some MCTD patients have also been diagnosed as bearing immune-mediated endothelial dysfunction and accelerated atherosclerosis. Therefore, endothelial dysfunction, which is an early marker of both vasculitis and premature atherosclerosis, may be exacerbated by those endothelium damaging antibodies present in MCTD patients.
Classical atherosclerosis accelerating factors, such as lipid abnormalities, were also observed in MCTD patients. The levels of total cholesterol and low-density lipoprotein cholesterol in MCTD patients were elevated, while the levels of high-density lipoprotein, apolipoprotein A1, and the natural antioxidant paraoxonase were clearly decreased [68, 69]. Increased C-reactive protein (CRP) levels and the presence of vascular endothelium damaging antibodies may have a direct effect on leukocyte recruitment and endothelial cell apoptosis, which might contribute to atherosclerosis in patients with MCTD [68, 70, 71]. Studies have indicated that antibodies to the family of heat-shock proteins (HSPs), for instance, the anti-HSP60 antibody, have an important effect on atherosclerosis by provoking vessel wall injury and result in early-onset atherosclerosis in MCTD patients [72].
Damage of the blood brain barrier (BBB) caused by thrombosis and vasculitis of small arachnoid arteries of the spinal cord may be the key link of myelopathy in MCTD [4]. Indeed, as an index of BBB permeability, the parameter Qalb increased in 8 of 14 (57%) patients, and appeared to inversely correlate with the anti-U1 RNP index [33]. Okada et al. also reported that 13 of 14 patients with aseptic meningitis presented with positive serum anti-U1 RNP antibodies, suggesting that anti-U1 RNP antibodies and the corresponding immune complex in cerebrospinal fluid (CSF) may function as pathogenic factors in NP involvements in SLE and MCTD [11, 15]. For patients with positive serum anti-U1 RNP antibodies, the sensitivity and specificity of CSF anti-U1 RNP antibodies for determining NP involvements were found to be 81.8% and 90.0%, respectively [33]. Therefore, BBB dysfunction significantly contributes to the pathogenesis of myelopathy in MCTD.
Clinical manifestations
Excessive damage to the spinal cord is the fundamental clinical manifestation of myelopathy in MCTD, and this damage progressively aggravates within several hours to weeks, and even months. Normally, a short period of nonspecific prodromal symptoms can be followed by bilateral lower limb paresthesia, accompanied by the sensation of numbness and reduction in muscular power [45,46,47,48]. These symptoms include fever, malasia, dizziness, nausea, vomiting, neck pain, and back or girdle pain of the trunk [45,46,47,48]. Progressively, the clinical manifestation evolves into a complete or partial paraplegia, or less frequently, tetraplegia, accompanied by sensory impairment, retention of urine, and fecal incontinence.
Motor dysfunction in patients with myelopathy is usually bilateral, but not necessarily symmetrical. Patients have been found to have multiple levels of motor dysfunction, such as complete paralysis, various levels of paresis, and a minor reduction in muscular power [44, 46,47,48,49]. In addition, patients with prominent gray matter signs can be clinically distinguished from those with white matter involvement. Usually, lower motor neuron syndrome with flaccidity and hyporeflexia is presented in patients with gray matter involvement. The symptom onset of motor dysfunction can be vigorous and is inclined to persist a long period with little or no recovery, which might reflect a possible irreversible injury to the medulla. Fever and urinary retention prior to irreversible paraplegia have been found to be associated with high disease activity [4, 47, 48]. Therefore, the signs of fever and urinary retention should be alerted in MCTD patients. The prompt diagnosis of this subtype is mandatory, but symptoms might be misrecognized due to the nature of the complexity of the disease. White matter myelopathy mainly manifests as upper-motor neuron syndrome with spasticity and hyperreflexia. Myelopathy with white matter involvement is less serious, and the course is more indolent with preserved strength. Intriguingly, the white matter involvement group is more often associated with optic neuritis. The prognosis is relatively good, although a higher risk of recurrence exists.
Consistent with the most commonly involved spinal segment, sensory impairment is usually found in the thoracic section. The area of sensory impairment is usually clearly limited to the same spinal segment indicated by motor dysfunction. Similarly, sensory impairment can also be identified with various intensities, which range from a complete lack of sensitivity to a selective analgesia and thermesthesia with preserved deep and vibratory sensibility [43, 44, 46,47,48,49].
Almost all patients with myelopathy in MCTD have autonomous nervous system dysfunction. Urine retention and intestinal peristalsis with the retention of gases and feces are the initial symptoms [46,47,48]. Progressively, urinary incontinence develops, and intestinal peristalsis returns, but fecal incontinence persists [4, 44]. Hence, patients gradually develop neurogenic bladder. Moreover, followed by trophic changes in paralyzed limbs, vasomotor dysfunction and perspiration disorders occur below the border of the region of sensory impairment. Subsequently, the limbs become cool and livid, and distal edema may appear.
Laboratory tests
Serologically, the presence of high titers of antinuclear antibodies (ANA) with a mottled pattern and antibodies targeting the U1-RNP (usually the IgG isotype) are sine qua non for the diagnosis of MCTD. Moreover, other antibodies, such as antidoublestranded deoxyribonucleic acid antibodies, antism antibodies, anti-aPL antibodies, and antiribonucleoprotein antibodies, can also be tested to support the diagnosis [4, 43, 44, 46,47,48,49].
The frequently reported hematological features include low-grade anemia or leukopenia, and sometimes thrombocytopenia and hemolytic anemia with positive Coomb’s test [4, 47, 48]. In the acute stage, the increasing rate of erythrocyte sedimentation, elevated values of CRP, and reduced levels of complement constituent can also be observed [48].
Apart from these serological and hematological tests, CSF examination is another fundamental tool for diagnosis confirmation in myelopathy, and excluding possible infection. A variable finding in CSF may be identified, ranging from normality to pleocytosis with lymphocyte predominance, and an increase in protein content [4, 45, 46, 48]. Unfortunately, the results of the CSF general tests and sediment test, which demonstrated as pleocytosis, high protein levels, and low glucose levels, could not distinguish patients with myelopathy from patients with myelitis in the course of bacterial infections. Therefore, additional adequate culturing experiments are required to exclude possible infections.
Imaging
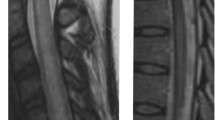
MRI is highly sensitive in evaluating injuries to the spinal cord. In clinical examinations, the protocol for MRI images is to cover the level corresponding to the clinically observed neurological damage. However, researchers strongly recommend this examination to include the entire cord. Inflammatory changes in the spinal cord are visible in T2weighted MRI [43, 45, 47]. With or without an edema, the intensification of a signal from the central part of the spinal cord can result in an increase in spinal cord thickness by several millimeters. As observed in several cases, the contrast uptake can be observed to determine the occurrence of spinal cord injuries [47]. Particularly, in patients with longer durations, these affected spinal cord segments may merely become thinner [49].
It is worth noting that MRI results may be normal in some cases, especially for patients in the early stages [46, 48]. The interpretation of MRI findings depends on the timing of the examination, as well as the stage of the disease. Another reason for this is possibly related to the utilized magnetic field strength. Hence, a magnetic field strength not lower than 1.5 T (Tesla units) is recommended to increase the yield. For patients with normal contrast MRI, a repeat examination at 2–7 days after the initial manifestation is recommended.
Computer tomography (CT) should not be used for the diagnosis of myelopathy due to its low sensitivity. This technology should only be used for cases where the MRI instrument is unavailable, and for the sake of excluding compressive causes of the spinal cord.
General diagnosis
Clues to the exact diagnosis of myelopathy in MCTD can be obtained from the CSF analysis, MRI examination, neurophysiological features, and immunological studies. Disease possibility should be considered when the MCTD patient progresses to one or more of the following signs/symptoms: sensory, motor, or autonomic dysfunction attributable to the spinal cord, and a clearly defined sensory level and peaking of symptoms within hours to several days, or even weeks, with or without bowel or bladder involvement. Furthermore, evidence of inflammation of the spinal cord is required, such as CSF pleocytosis, elevated protein content and IgG level, or MRI with gadolinium consistent with myelopathy. The diagnosis of myelopathy cannot be ruled out in patients without signs of any abnormalities of the spinal cord by MRI examination.
Differential diagnosis
Spinal cord transverse damage is resulted from multiple etiological causes [57, 73,74,75,76]. The key features giving most differential diagnostic value include speed of symptom onset (hyperacute, acute, subacute, or chronic), disease course (monophasic or relapsing or progressive, complete partial or no recovery, stable or fluctuating), lesion characteristics on MRI (the length and position, along with the cross-sectional pattern of involvement such as gray or white matter, anterior or posterior or lateral locations, symmetrical or asymmetrical and gadolinium enhancement pattern), and other additional clinical features. Myelopathy in MCTD usually manifests as acute or subacute course, so it is particularly important to differentiate it from diseases presenting as an acute or subacute onset (Fig. 1).

Flow diagram approach to MCTD patient presenting acute/subacute myelopathy. AQP4, aquaporin-4; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; NMO, neuromyelitis optica; CIS, clinically isolated syndrome; OCB, oligoclonal bands; MCTD, mixed connective tissue disease; LETM, longitudinal extensive transverse myelopathy; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging
In MCTD patients with abnormal CSF test results and fever, a possible concurrent neural infection must be considered. Since patients with diagnosed MCTD usually receive immunosuppressive agents, they are at higher risk of pathogenic infection. Therefore, the careful examination of the patient’s history and a thorough analysis of all clinical manifestations, especially additional adequate CSF culturing experiments are required to exclude possible infections. Acute to subacute infective myelitis is most commonly viral and detecting the viral DNA in the CSF may help.
The etiology of vascular myopathy mainly includes arteriovenous malformations, cavernomas, systemic hypoperfusion, vasculitis, and embolism [77]. The symptom onset is usually abrupt (within minutes), but the time from onset to nadir may be a few hours. The clinical symptom of spinal ischemia can cover many manifestations, such as dissociation of sensation, spastic paraparesis or tetraparesis, and the perseveration of proprioception, which are typical for vascular lesions of the spinal cord [78]. The MRI results are featured by pencil-like lesions in the anterior part of the spinal cord with typical hyper-intense long lesions. Diffuse weight imaging is promising in the diagnosis of spinal ischemia.
Notably, patients with myelopathy are inclined to be erroneously diagnosed as isolated nervous system diseases with multiple sclerosis (MS) and neuromyelitis optica (NMO) [75]. Therefore, the definite identification of these three diseases (NMO, MS, and myelopathy in MCTD) is particularly important (Table 3). As mentioned above, some patients may present with optic neuritis at the same time. In particular, for MCTD patients complicated with myelopathy who also have optic neuritis, other diseases, such as MS and NMO, should be excluded first, and then the simultaneous involvement of optic nerve and spinal cord should be considered.
MCTD patients with less than 3 spinal segments involved should be differentiated from MS or clinically isolated syndrome (CIS). The diagnosis of MS can be confirmed by past demyelination history combined with multiple demyelination lesions in brain and spinal cord MRI, and positive oligonucleotide band in CSF [84]. CIS refers to the first episode of isolated central nervous system demyelinating disease, which mainly includes optic neuritis, isolated brain stem damage, or spinal cord injury [82, 85]. CIS of the spinal cord is featured with dominant and asymmetrical sensory signs and symptoms, including mostly paraesthesias which usually slowly spread from the legs to the upper part of the body, are more pronounced on one side, and often presents with a band-like sensation, or pressure around the abdomen or chest [85]. MRI in patients with CIS is also typical with small demyelinating lesions, which involve less than two segments, and are most often located in the posterior or lateral part of the spinal cord [82]. Acute or subacute onset with main cervical spinal cord involvement and incomplete spinal cord injury involving less than two segments in MRI can help confirm the diagnosis of spinal cord CIS. Particularly, for patients who have experienced CIS and have an abnormal MRI scan, a second episode (or relapse) would appear, and they would manifest the onset of clinically definite MS.
Another diagnostic problem is posed by MCTD patients who manifest as LETM. Of note, LETM can be the first presentation of NMO, and thus all such patients need to have their serology for AQP4-Ab and myelin oligodendrocyte glycoprotein (MOG) antibody tested [74, 86]. Recent studies have confirmed that AQP4 antibodies and NMO-IgG are highly specific and sensitive in NMO patients, making them part of the diagnostic criteria for NMO [79]. Its clinical features are recurrent optic neuritis and transverse spinal cord injury. The typical imaging findings are bright spotty lesions on T2-weighted images and corresponding dark lesions on T1-weighted images with central gray matter or holocord involvement, usually including the thoracic cord [79, 87]. However, AQP4-Ab positive NMO can present with short lesions in up to 14% of initial TM attacks [88]. The lesions can be eccentric, and some are asymptomatic. Ring-enhancing lesions develop in about one-third of these patients [80]. These imaging features have been described as relatively specific for distinguishing NMO from other entities, including MS.
Sporadically reported cases suggest that NMO may coexist with SLE, SS, MCTD, myasthenia gravis, autoimmune thyroiditis, or other systemic autoimmune diseases [81, 89,90,91,92]. Studies conducted by Arabshahi B et al. revealed that about half of NMO patients with systemic connective tissue disease had positive serum AQP4-Ab, while the serum AQP4-Ab in SLE or systemic sclerosis patients without NMO were negative [93]. MCTD patients complicated with spinal cord white matter damage and optic neuritis and positive AQP4-Ab should be considered as cases of MCTD combined with NMO, rather than the basic pathological changes of systemic autoimmune diseases, such as nervous system complications caused by vasculitis.
For MCTD patients with LETM, clinicians may not exclude a metabolic cause. The most common metabolic myelopathy is caused by vitamin B12 deficiency [94]. Of note, the neurological disease may occur either with hematological manifestations or as a result of a functional deficiency without decreased serum vitamin B12 level, which is supported by a raised serum methylmalonic acid or homocysteine and a low concentration of transcobalamin-2 [94]. The classic MRI finding is a long cord lesion with symmetrical T2 hyperintensity in the posterior and lateral columns, which most commonly involves the thoracic cord [95]. Anterior column T2 hyperintensity and contrast enhancement of the lesion are rare but can occur in isolated cases. Furthermore, copper deficiency is another cause of metabolic myelopathy. The differences in imaging of copper deficiency cases, compared with vitamin B12 deficiency, include increased prevalence of cervical cord and central cord involvement in addition to the similar posterior column pathology [96]. A low serum copper and ceruloplasmin would be in keeping with this diagnosis.
Treatment
The optimal therapeutic strategy of myelopathy in MCTD is less well-defined, and there is no unanimous therapy due to its rarity. In order to minimize permanent neurological damage to the cord, an aggressive approach with the prompt treatment of high-dose steroids and immunosuppressors is recommended, as referred by the treatment scheme of myelopathy in SLE [57, 58, 73]. The treatment was initiated with pulse dose therapy by intravenous injection of 1 g/day of methylprednisolone for 3 days, combined with azathioprine at a dose of 1–2 mg/kg per day [47, 48]. On the 4th day after treatment initiation, azathioprine and steroids were subsequently changed to oral prednisone at 1 mg/kg bodyweight/day for 6 months to 1 year, and the prednisone dosage was slowly tapered to a maintenance dose at 10 mg per day in 3 months. However, the duration of maintenance treatment would vary in patients.
Some scholars suggest that patients in critical condition, such as poor responsiveness to hormone therapy or recurrence, can be treated with intravenous immunoglobulin injection [43, 47]. In addition, in order to rapidly clear specific autoantibodies in plasma and prevent the formation of permanent injury to the spinal cord, plasmapheresis can be conducted, although the responses vary in patients [43, 47]. Bhinder et al. [47] reported a severe case with monthly IVIG at a dosage of 0.4 g/kg per day for 5 days, which was continued for a period of 6 months apart from the high-dose steroid and immunosuppressors. After treatment, the patient’s condition significantly improved, and there was no relapse during the follow-up. Flenchter et al. [43] reported a case with recurrent episodes of optic neuropathy and transverse myelopathy. At the time of the second attack, plasmapheresis performed at regular intervals of 3 days while immunosuppressive medication (azathioprine, prednisolone) was continued. The patient’s symptoms significantly improved after the 6th plasmapheresis, and the time interval of the plasmapheresis was extended to approximately once a month. Thereafter, each recurrence was well-controlled with the combination of plasmapheresis and immunosuppressive medication.
In addition, some studies have also reported the use of rituximab (anti-CD20 receptor antibody), autologous bone marrow transplantation, intrathecal dexamethasone, and methotrexate to treat severe SLE neurological complications [26, 65, 97]. When the patient has contraindications, or does not respond to hormone and immunosuppressive therapy, the above treatment plans can be considered. Moreover, hydroxychloroquine would reduce the number of relapses in SLE patients [60, 98].
Symptomatic treatment is critical in the therapy of patients with ATM. When urinary retention occurs, Foley catheter insertion to the urinary bladder is necessary in the early stage of the disease, because the overflow of the bladder caused by sphincter paralysis may lead to rupture [48]. Patients with spinal paralysis can easily develop bedsores due to immobility, sensory impairment, and damaged autonomic nervous system. Therefore, intensive anti-bedsore care is needed. Patients with myelopathy also require complex and intensive rehabilitation, which should be started at the diagnosis of the disease. Before the start of the rehabilitation training, venous thrombosis of the limbs needs to be excluded to avoid serious consequences.
Prognosis
The clinical course of myelopathy in MCTD varies. The prognosis of patients can be obvious improvement with minor sequelae, partial remissions, arrest of progress of the disease and deterioration of neurological symptoms, and sometimes relapse or death might occur [4, 43,44,45,46,47,48,49]. The time needed for improvements in neurological function may vary, which ranges between several days and many months [46,47,48,49]. It has recently been agreed that the early introduction of immunosuppressive therapy with intravenous azathioprine and high doses of glucocorticosteroids, followed by oral low doses of glucocorticosteroids, improves the long-term prognosis of patients with myelopathy in MCTD.
As highlighted by previous reports, serious neurological manifestations, such as myelopathy, may rarely complicate in the course of MCTD, but these appear to intrinsically have a better prognosis than a similar presentation in SLE. In addition, the prognosis of patients with MRI alterations is worse than that of patients without such abnormalities [43, 47]. The prognosis is particularly poor in patients with long extensive spinal lesions of the spinal cord [43]. When patients have obvious improvement at the onset of immunosuppressive therapy, these patients would probably also have a good prognosis, as they would respond well to the immunosuppressive therapy.
Flenchter et al. [43] reported a patient with myelopathy in MCTD, who received long-term, low-dose hormone therapy. One of the recurrences in this patient was observed right after hormone dosage reduction. In order to prevent further recurrences, a continuous maintenance immunosuppressive treatment was mandatory, as suggested by this case. In addition, hormone reduction in the patients receiving long-term low-dose hormone therapy should be performed with caution, and regular laboratory examinations should be periodically conducted.
Conclusion
Myelopathy in MCTD is extremely rare. Although its diagnosis is complex due to the heterogeneity of the pathological processes, its treatment remains challenging. Similar to MS and SLE complicated by myelopathy, this disease often demonstrates spinal cord involvement with serious neurological complications, such as paralysis or muscular paresis, sensory impairment, and smooth muscle dysfunction. Progressive clinical manifestations combined with laboratory examinations and MRI examinations play important roles in the diagnosis of myelopathy in MCTD. CSF examination is a fundamental tool for diagnosis confirmation in myelopathy, and excluding possible infection. Although the treatment for myelopathy in MCTD might be individualized, an aggressive approach with prompt treatment of high-dose steroid and immunosuppressors is recommended to minimize permanent neurological damage to the spinal cord, and plasmapheresis can also be conducted to rapidly clear the specific autoantibodies in the plasma. The oral administration of low doses of glucocorticosteroids and other immunosuppressors can improve the long-term prognosis of patients with myelopathy in MCTD. The ongoing surveillance and more reports on this rare presentation in MCTD would greatly help in better understanding its pathomechanisms, and shed more light on the definite diagnosis and effective therapy of this disease.
References
Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR (1972) Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med 52(2):148–159
Hoffman R, Bezruczko N, Perkins K (2012) An external validation study of a classification of mixed connective tissue disease and systemic lupus erythematosus patients. J Appl Meas 13(2):205–216
Perkins K, Hoffman RW, Bezruczko N (2008) A Rasch analysis for classification of systemic lupus erythematosus and mixed connective tissue disease. J Appl Meas 9(2):136–150
Weiss T, Nelson J, Woolsey R, Zuckner J, Baldassare A (1978) Transverse myelitis in mixed connective tissue disease. Arthritis Rheum 21(8):982–986
Nichtweiß M, Weidauer S (2015) Differential diagnosis of acute myelopathies: an update. Clin Neuroradiol 25(Suppl 2):183–187
Minkin W, Rabhan N (1976) Mixed connective tissue disease. Arch Dermatol 112(11):1535–1538
Gunnarsson R, Molberg O, Gilboe IM, Gran JT (2011) The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis 70(6):1047–1051. https://doi.org/10.1136/ard.2010.143792
Ungprasert P, Crowson CS, Chowdhary VR, Ernste FC, Moder KG, Matteson EL (2016) Epidemiology of mixed connective tissue disease, 1985–2014: a population-based study. Arthritis Care Res 68(12):1843–1848. https://doi.org/10.1002/acr.22872
Ferucci E, Johnston J, Gordon C, Helmick C, Lim S (2017) Prevalence of mixed connective tissue disease in a population-based Registry of American Indian/Alaska Native People in 2007. Arthritis Care Res 69(8):1271–1275
Paradowska-Gorycka A, Stypinska B, Olesinska M, Felis-Giemza A, Manczak M, Czuszynska Z, Zdrojewski Z, Wojciechowicz J, Jurkowska M (2016) Association of HLA-DRB1 alleles with susceptibility to mixed connective tissue disease in Polish patients. Hla 87(1):13–18. https://doi.org/10.1111/tan.12698
Paradowska-Gorycka A (2015) U1-RNP and Toll-like receptors in the pathogenesis of mixed connective tissue diseasePart II. Endosomal TLRs and their biological significance in the pathogenesis of mixed connective tissue disease. Reumatologia 53(3):143–151
Hameenkorpi R, Ruuska P, Forsberg S, Tiilikainen R, Makitalo R, Hakala M (1993) More evidence of distinctive features of mixed connective tissue disease. Scand J Rheumatol 22(2):63–68
Yoshida K, Inoue H, Komai K, Yamane T, Hashiramoto A, Shiozawa K, Shiozawa S (2013) Mixed connective tissue disease is distinct from systemic lupus erythematosus: study of major histocompatibility complex class I polypeptide-related sequence A and HLA gene polymorphisms. Tissue Antigens 81(1):44–45. https://doi.org/10.1111/tan.12027
Martínez-Barrio J, Valor L, López-Longo F (2018) Facts and controversies in mixed connective tissue disease. Med Clin (Barc) 150(1):26–32
Dima A, Jurcut C, Baicus C (2018) The impact of anti-U1-RNP positivity: systemic lupus erythematosus versus mixed connective tissue disease. Rheumatol Int 38(7):1169–1178. https://doi.org/10.1007/s00296-018-4059-4
Sharp GC, Irvin WS, May CM, Holman HR, McDuffie FC, Hess EV, Schmid FR (1976) Association of antibodies to ribonucleoprotein and Sm antigens with mixed connective-tissue disease, systematic lupus erythematosus and other rheumatic diseases. N Engl J Med 295(21):1149–1154. https://doi.org/10.1056/nejm197611182952101
Minkin W, Rabhan N (1977) Mixed connective tissue disease. Arch Intern Med 137(10):1484
Hoffmann-Vold AM, Gunnarsson R, Garen T, Midtvedt Ø, Molberg Ø (2015) Performance of the 2013 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Systemic Sclerosis (SSc) in large, well-defined cohorts of SSc and mixed connective tissue disease. J Rheumatol 42(1):60–63
Doria A, Ghirardello A, de Zambiasi P, Ruffatti A, Gambari PF (1992) Japanese diagnostic criteria for mixed connective tissue disease in Caucasian patients. J Rheumatol 19(2):259–264
Amigues JM, Cantagrel A, Abbal M, Mazieres B (1996) Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. Autoimmunity Group of the Hospitals of Toulouse. J Rheumatol 23(12):2055–2062
Cappelli S, Bellando Randone S, Martinovic D, Tamas MM, Pasalic K, Allanore Y, Mosca M, Talarico R, Opris D, Kiss CG, Tausche AK, Cardarelli S, Riccieri V, Koneva O, Cuomo G, Becker MO, Sulli A, Guiducci S, Radic M, Bombardieri S, Aringer M, Cozzi F, Valesini G, Ananyeva L, Valentini G, Riemekasten G, Cutolo M, Ionescu R, Czirjak L, Damjanov N, Rednic S, Matucci Cerinic M (2012) "To be or not to be," ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum 41(4):589–598. https://doi.org/10.1016/j.semarthrit.2011.07.010
Flåm S, Gunnarsson R, Garen T, Lie B, Molberg Ø (2015) The HLA profiles of mixed connective tissue disease differ distinctly from the profiles of clinically related connective tissue diseases. Rheumatology 54(3):528–535
Lage LV, Caleiro MT, Carvalho JF (2010) Proposed disease activity criteria for mixed connective tissue disease. Lupus 19(2):223–224. https://doi.org/10.1177/0961203309345782
Rayes HA, Al-Sheikh A, Al Dalaan A, Al Saleh S (2002) Mixed connective tissue disease: the King Faisal Specialist Hospital experience. Ann Saudi Med 22(1–2):43–46
Habets WJ, de Rooij DJ, Salden MH, Verhagen AP, van Eekelen CA, van de Putte LB, van Venrooij WJ (1983) Antibodies against distinct nuclear matrix proteins are characteristic for mixed connective tissue disease. Clin Exp Immunol 54(1):265–276
Xuan Z, Yi D, Fu-Lin T, Fen-Chun Z (1999) Central nervous system involvement in systemic lupus erythematosus in a hospital-based study of 171 cases: the possible therapeutic role of intrathecal therapy. J Clin Rheumatol 5(6):314–319
Bennett RM, Bong DM, Spargo BH (1978) Neuropsychiatric problems in mixed connective tissue disease. Am J Med 65(6):955–962
Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC (1999) Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum 42(5):899–909. https://doi.org/10.1002/1529-0131(199905)42:5<899::aid-anr8>3.0.co;2-l
Hojaili B, Barland P (2006) Trigeminal neuralgia as the first manifestation of mixed connective tissue disorder. J Clin Rheumatol 12(3):145–147. https://doi.org/10.1097/01.rhu.0000222045.70861.a5
Debette S, Germain DP (2014) Neurologic manifestations of inherited disorders of connective tissue. Handb Clin Neurol 119:565–576. https://doi.org/10.1016/b978-0-7020-4086-3.00037-0
Nadeau SE (2002) Neurologic manifestations of connective tissue disease. Neurol Clin 20(1):151–178 vi
Karmacharya P, Mainali N, Aryal M, Lloyd B (2013) Recurrent case of ibuprofen-induced aseptic meningitis in mixed connective tissue disease. BMJ Case Rep 2013. https://doi.org/10.1136/bcr-2013-009571
Sato T, Fujii T, Yokoyama T, Fujita Y, Imura Y, Yukawa N, Kawabata D, Nojima T, Ohmura K, Usui T, Mimori T (2010) Anti-U1 RNP antibodies in cerebrospinal fluid are associated with central neuropsychiatric manifestations in systemic lupus erythematosus and mixed connective tissue disease. Arthritis Rheum 62(12):3730–3740. https://doi.org/10.1002/art.27700
Shah P, Dhakre V, Bhabhor A, Prasad A (2018) Superior sagittal sinus thrombosis in a case of mixed connective tissue disease. BMJ Case Rep 2018. https://doi.org/10.1136/bcr-2018-225078
Kim ST, Brinjikji W, Lanzino G, Kallmes DF (2016) Neurovascular manifestations of connective-tissue diseases: a review. Interv Neuroradiol 22(6):624–637. https://doi.org/10.1177/1591019916659262
Matsui H, Udaka F, Oda M, Kubori T, Nishinaka K, Kameyama M (2006) Encephalopathy and severe neuropathy due to probable systemic vasculitis as an initial manifestation of mixed connective tissue disease. Neurol India 54(1):83–85
Leibfarth J, Persellin R (1976) Characteristics of patients with serum antibodies to extractable nuclear antigens. Arthritis Rheum 19(5):851–856
Hajas A, Szodoray P, Barath S, Sipka S, Rezes S, Zeher M, Sziklai I, Szegedi G, Bodolay E (2009) Sensorineural hearing loss in patients with mixed connective tissue disease: immunological markers and cytokine levels. J Rheumatol 36(9):1930–1936
Kiani I, Qureshi S, Shah F (2014) Depression and seizures as the main neuropsychiatric manifestation of mixed connective tissue disorder. J Coll Physicians Surg Pak 24(Suppl 2):S141–S143
Manohar H, Kuppili P, Menon V (2017) Major depression: An under reported neuropsychiatric manifestation of mixed connective tissue disease. Asian J Psychiatr 30:54–55
Nowicka-Sauer K, Czuszynska Z, Majkowicz M, Smolenska Z, Jarmoszewicz K, Olesinska M, Siebert J (2012) Neuropsychological assessment in mixed connective tissue disease: comparison with systemic lupus erythematosus. Lupus 21(9):927–933
Laffon-Pioger M, Rocher F, Cohen M, Chanalet S, Thomas P, Lebrun C (2010) Bilateral optic neuropathy with loss of vision after an influenza vaccination in a patient suffering from mixed connective tissue disease. Rev Neurol (Paris) 166(12):1024–1027
Flechtner K, Baum K (1994) Mixed connective tissue disease: recurrent episodes of optic neuropathy and transverse myelopathy. Successful treatment with plasmapheresis. J Neurol Sci 126(2):146–148
Pedersen C, Bonen H, Boesen F (1987) Transverse myelitis in mixed connective tissue disease. Clin Rheumatol 6(2):290–292
Obara K, Tanaka K (1991) A case of mixed connective tissue disease (MCTD) associated with transverse myelitis responding to pulse therapy. Rinsho Shinkeigaku 31(11):1197–1201
Mok CC, Lau CS (1995) Transverse myelopathy complicating mixed connective tissue disease. Clin Neurol Neurosurg 97(3):259–260
Bhinder S, Harbour K, Majithia V (2007) Transverse myelitis, a rare neurological manifestation of mixed connective tissue disease--a case report and a review of literature. Clin Rheumatol 26(3):445–447. https://doi.org/10.1007/s10067-005-0158-1
Hao Y, Feng L, Teng Y, Cheng Y, Feng J (2018) Management of multiple neurological complications in mixed connective tissue disease: a case report. Medicine 97(31):e11360. https://doi.org/10.1097/md.0000000000011360
Weatherby SJ, Davies MB, Hawkins CP, Haq N, Dawes P (2000) Transverse myelopathy, a rare complication of mixed connective tissue disease: comparison with SLE related transverse myelopathy. J Neurol Neurosurg Psychiatry 68(4):532–533
Kucuk A, Uslu AU, Yilmaz R, Salbas E, Solak Y, Tunc R (2017) Relationship between prevalence and severity of restless legs syndrome and anemia in patients with systemic lupus erythematosus. Int J Rheum Dis 20(4):469–473. https://doi.org/10.1111/1756-185x.12793
Schedel J, Kuchenbuch S, Schoelmerich J, Feuerbach S, Geissler A, Mueller-Ladner U (2010) Cerebral lesions in patients with connective tissue diseases and systemic vasculitides: are there specific patterns? Ann N Y Acad Sci 1193:167–175
Hetlevik S, Flatø B, Rygg M, Nordal E, Brunborg C, Hetland H, Lilleby V (2017) Long-term outcome in juvenile-onset mixed connective tissue disease: a nationwide Norwegian study. Ann Rheum Dis 76(1):159–165
Rahmanzadeh R, Rahmanzade R, Zabihiyeganeh M (2016) Posterior reversible encephalopathy syndrome in a patient with mixed connective tissue disease: a case report. J Med Case Rep 10(1):145
Tristano A (2009) Autoimmune diseases associated with transverse myelitis. Review. Investig Clin 50(2):251–270
Nived O, Sturfelt G, Liang MH, De Pablo P (2003) The ACR nomenclature for CNS lupus revisited. Lupus 12(12):872–876. https://doi.org/10.1191/0961203303lu495oa
Kovacs B, Lafferty TL, Brent LH, DeHoratius RJ (2000) Transverse myelopathy in systemic lupus erythematosus: an analysis of 14 cases and review of the literature. Ann Rheum Dis 59(2):120–124
Ahn S, Hong S, Lim D, Ghang B, Kim Y, Lee C, Yoo B (2018) Clinical features and prognoses of acute transverse myelitis in patients with systemic lupus erythematosus. Korean J Intern Med. 34(2):442-451. doi: 10.3904/kjim.2016.383.
Chiganer E, Hryb J, Carnero Contentti E (2017) Myelitis and lupus: clinical manifestations, diagnosis and treatment. Review Reumatol Clin 13(6):344–348
Zahid A, Mubashir A, Mirza S, Naqvi I, Talib A (2018) Systemic lupus erythematosus presenting as longitudinally extensive transverse myelitis and nephritis: a case report. Cureus 10(4):e2402
Saison J, Costedoat-Chalumeau N, Maucort-Boulch D, Iwaz J, Marignier R, Cacoub P, Vital-Durand D, Hot A, Tebib J, Aumaitre O, Schleinitz N, Sarrot-Reynauld F, Broussolle C, Seve P (2015) Systemic lupus erythematosus-associated acute transverse myelitis: manifestations, treatments, outcomes, and prognostic factors in 20 patients. Lupus 24(1):74–81. https://doi.org/10.1177/0961203314547795
Harel L, Sandborg C, Lee T, von Scheven E (2006) Neuropsychiatric manifestations in pediatric systemic lupus erythematosus and association with antiphospholipid antibodies. J Rheumatol 33(9):1873–1877
Proposed diagnostic criteria and nosology of acute transverse myelitis (2002). Neurology 59 (4):499–505
Flores-Silva F, Longoria-Lozano O, Aguirre-Villarreal D, Sentíes-Madrid H, Vega-Boada F, Díaz de León-Sánchez E, Murra-Antón S, Morales-Moreno S, Quintanilla-González L, Fragoso-Loyo H, Guraieb-Chaín P, Higuera-Calleja J, Ceballos-Ceballos J, Treviño-Frenk I, González-Duarte A, Dávila-Maldonado L, Cantú-Brito C, Valdés-Ferrer S (2018) Natural history of longitudinally extensive transverse myelitis in 35 Hispanic patients with systemic lupus erythematosus: good short-term functional outcome and paradoxical increase in long-term mortality. Lupus 27(8):1279–1286
Chan K, Boey M (1996) Transverse myelopathy in SLE: clinical features and functional outcomes. Lupus 5(4):294–299
Lehnhardt F, Scheid C, Holtik U, Burghaus L, Neveling M, Impekoven P, Rüger A, Hallek M, Jacobs A, Rubbert A (2006) Autologous blood stem cell transplantation in refractory systemic lupus erythematodes with recurrent longitudinal myelitis and cerebral infarction. Lupus 15(4):240–243
Sherer Y, Hassin S, Shoenfeld Y, Levy Y, Livneh A, Ohry A, Langevitz P (2002) Transverse myelitis in patients with antiphospholipid antibodies--the importance of early diagnosis and treatment. Clin Rheumatol 21(3):207–210
Soltesz P, Bereczki D, Szodoray P, Magyar M, Der H, Csipo I, Hajas A, Paragh G, Szegedi G, Bodolay E (2010) Endothelial cell markers reflecting endothelial cell dysfunction in patients with mixed connective tissue disease. Arthritis Res Ther 12(3):R78
Haładyj E, Paradowska-Gorycka A, Felis-Giemza A, Olesińska M (2016) Immunity and early atherosclerosis in the course of systemic lupus erythematosus, mixed connective tissue disease and antiphospholipid syndrome. Reumatologia 54(4):187–195
Hajas A, Sandor J, Csathy L, Csipo I, Barath S, Paragh G, Seres I, Szegedi G, Shoenfeld Y, Bodolay E (2011) Vitamin D insufficiency in a large MCTD population. Autoimmun Rev 10(6):317–324
Svenungsson E, Cederholm A, Jensen-Urstad K, Fei G, de Faire U, Frostegård J (2008) Endothelial function and markers of endothelial activation in relation to cardiovascular disease in systemic lupus erythematosus. Scand J Rheumatol 37(5):352–359
Bakri Hassan A, Rönnelid J, Gunnarsson I, Karlsson G, Berg L, Lundberg I (1998) Increased serum levels of immunoglobulins, C-reactive protein, type 1 and type 2 cytokines in patients with mixed connective tissue disease. J Autoimmun 11(5):503–508
Bodolay E, Prohászka Z, Paragh G, Csipő I, Nagy G, Laczik R, Demeter N, Zöld E, Nakken B, Szegedi G, Szodoray P (2014) Increased levels of anti-heat-shock protein 60 (anti-Hsp60) indicate endothelial dysfunction, atherosclerosis and cardiovascular diseases in patients with mixed connective tissue disease. Immunol Res 60(1):50–59
Tristano AG (2009) Autoimmune diseases associated with transverse myelitis. Rev Investig Clin 50(2):251–270
Habek M, Adamec I, Pavliša G, Brinar V (2012) Diagnostic approach of patients with longitudinally extensive transverse myelitis. Acta Neurol Belg 112(1):39–43
Kayal A, Goswami M, Das M, Basumatary L, Bhowmick S, Synmon B (2017) Etiological profile of noncompressive myelopathies in a tertiary care hospital of Northeast India. Ann Indian Acad Neurol 20(1):41–50
Wang F, Guo D, Liu Z, Zhou A, Wei C, Jia J (2018) Neurosarcoidosis: clinical characteristics, diagnosis, and treatment in eight Chinese patients. Neurol Sci 39(10):1725–1733. https://doi.org/10.1007/s10072-018-3491-2
Novy J, Carruzzo A, Maeder P, Bogousslavsky J (2006) Spinal cord ischemia: clinical and imaging patterns, pathogenesis, and outcomes in 27 patients. Arch Neurol 63(8):1113–1120
Masson C, Pruvo J, Meder J, Cordonnier C, Touzé E, De La Sayette V, Giroud M, Mas J, Leys D (2004) Spinal cord infarction: clinical and magnetic resonance imaging findings and short term outcome. J Neurol Neurosurg Psychiatry 75(10):1431–1435
Vaknin-Dembinsky A, Karussis D, Avichzer J, Abramsky O (2014) NMO spectrum of disorders: a paradigm for astrocyte-targeting autoimmunity and its implications for MS and other CNS inflammatory diseases. J Autoimmun 54:93–99
NL Z, PP M, BG W, CF L, Y G SJP, KN K, TJ K, DM W, N K EPF (2017) Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 88(3):218–225
Ogaki K, Hirayama T, Chijiiwa K, Fukae J, Furuya T, Noda K, Fujishima K, Hattori N, Takahashi T, Okuma Y (2012) Anti-aquaporin-4 antibody-positive definite neuromyelitis optica in a patient with thymectomy for myasthenia gravis. Neurologist 18(2):76–79
Metz L, Li D, Traboulsee A, Duquette P, Eliasziw M, Cerchiaro G, Greenfield J, Riddehough A, Yeung M, Kremenchutzky M, Vorobeychik G, Freedman M, Bhan V, Blevins G, Marriott J, Grand'Maison F, Lee L, Thibault M, Hill M, Yong V (2017) Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med 376(22):2122–2133
Dehghani A, Nayeri N, Ebadi A (2017) Development and validation of the coping with multiple sclerosis questionnaire. Mult Scler Relat Disord 18:49–55
Axisa P, Hafler D (2016) Multiple sclerosis: genetics, biomarkers, treatments. Curr Opin Neurol 29(3):345–353
Kitzler H, Wahl H, Eisele J, Kuhn M, Schmitz-Peiffer H, Kern S, Rutt B, Deoni S, Ziemssen T, Linn J (2018) Multi-component relaxation in clinically isolated syndrome: lesion myelination may predict multiple sclerosis conversion. Neuroimage Clin 20:61–70
Jacob A, McKeon A, Nakashima I, Sato D, Elsone L, Fujihara K, de Seze J (2013) Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry 84(8):922–930
Zhou C, He Y, Gao X, Zhu Y, Chao Z, Wang X (2018) Neuromyelitis optical spectrum disorders presenting with isolated “inverted V” sign in area postrema. Neurol Sci 39(7):1299–1301. https://doi.org/10.1007/s10072-018-3302-9
EP F, BG W, KN K, VA L, CF L, A M DMW, EA S, Y J ESH, SJ P (2015) Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA neurology 72(1):81–87
Zhong YH, Zhong ZG, Zhou Z, Ma ZY, Qiu MY, Peng FH, Zhang WX (2017) Comparisons of presentations and outcomes of neuromyelitis optica patients with and without Sjogren's syndrome. Neurol Sci 38(2):271–277. https://doi.org/10.1007/s10072-016-2751-2
Squatrito D, Colagrande S, Emmi L (2010) Devic's syndrome and primary APS: a new immunological overlap. Lupus 19(11):1337–1339
Mehta L, Samuelsson M, Kleiner A, Goodman A, Anolik J, Looney R, Schwid S (2008) Neuromyelitis optica spectrum disorder in a patient with systemic lupus erythematosus and anti-phospholipid antibody syndrome. Mult Scler 14(3):425–427
Kister I, Gulati S, Boz C, Bergamaschi R, Piccolo G, Piccolo G, Oger J, Swerdlow M (2006) Neuromyelitis optica in patients with myasthenia gravis who underwent thymectomy. Arch Neurol 63(6):851–856
Arabshahi B (2006) Devic disease in a child with primary Sjogren syndrome. J Child Neurol 21(4):284–286
RN S (2018) Metabolic and toxic myelopathies. Continuum (Minneapolis, Minn) 24(2):427–440
AH K, A M GB (2000) Subacute combined degeneration of the spinal cord with involvement of the anterior columns: a new MRI finding. Neuroradiology 42(2):115–117
N K JEA, CJ K, JD P (2006) Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology 48(2):78–83
Armstrong D, McCarron M, Wright G (2006) SLE-associated transverse myelitis successfully treated with Rituximab (anti-CD20 monoclonal antibody). Rheumatol Int 26(8):771–772
Klaiman MD, Miller SD (1993) Transverse myelitis complicating systemic lupus erythematosus: treatment including hydroxychloroquine. Case report. Am J Phys Med Rehabil 72(3):158–161
Acknowledgments
The authors thank Dr. Mingqin Zhu for her guidance and support to this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hao, Y., Xin, M., Wang, S. et al. Myelopathy associated with mixed connective tissue disease: clinical manifestation, diagnosis, treatment, and prognosis. Neurol Sci 40, 1785–1797 (2019). https://doi.org/10.1007/s10072-019-03935-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-019-03935-y