
Abstract
Neurosarcoidosis is relatively rare and has diverse manifestations. The clinical characteristics, diagnosis, treatment, and outcome for neurosarcoidosis in China are poorly understood. We retrospectively analyzed the clinical features, laboratory and imaging results, treatment, and outcomes in patients who met the criteria for definite or probable neurosarcoidosis in Xuan Wu Hospital of Capital Medical University from 2000 to 2015. Eight patients were included in this study, accounting for 5.84% of all cases with sarcoidosis. The mean age at onset was 50.25 years, and 75% of the patients were female. Five cases had a prior diagnosis of extraneurologic sarcoidosis, leading to a shorter lag time between onset of symptoms and diagnosis (3.4 vs. 16.2 months). Neurological symptoms were the first clinical feature of sarcoidosis in three cases, and no patients presented isolated nervous system manifestation. The most common symptom was sensory disturbance, and the most common site of nervous system involvement was brain parenchyma and meninges. Disturbance of consciousness, seizures, hydrocephalus, and abnormal CSF assays were associated with poor prognosis. All patients were treated with corticosteroids and one was also given azathioprine. Five patients had complete or partial improvement, one remained stabilized, and two deteriorated and died. Neurosarcoidosis is difficult to diagnose early and might be associated with a poor prognosis. Tissue biopsy for a definitive diagnosis and aggressive therapy with corticosteroids plus other alternative immunosuppressive treatment should be recommended in China.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis is a multisystem, non-caseating granulomatous disease [1, 2]. Its pathogenesis is generally considered to be related to the abnormal immune response induced by both infectious and non-infectious factors in susceptible individuals [3]. The incidence rate of sarcoidosis is highest in northern European and African-American populations [1, 2, 4]. However, it is relatively low in Asia, such as Japan and Singapore [5, 6]. It occurs more commonly in adults within 20–40 years old [2] and usually affects the lung, skin, eyes, and liver.
Previous clinical studies revealed that approximately 5–15% of patients with sarcoidosis had neurological involvement [3, 7,8,9,10]. However, this rate is much higher at autopsy, up to 25%, suggesting that there are a large number of patients with subclinical neurosarcoidosis [3, 7]. Neurosarcoidosis may involve any part of the nervous system, including cranial nerves, brain parenchyma, meninges, spinal cord, and peripheral neuropathy. Therefore, it has diverse manifestations and different courses [11]. If sarcoidosis exclusively affects the nervous system, or neurological manifestations appear in the patients with inactive systemic sarcoidosis, diagnosis is much more challenging to make, and histological evidence of non-caseating granulomas of the nervous system tissue is needed [12].
There have been no accepted treatment guidelines for neurosarcoidosis so far, and most recommendations are based on the results of case observations and expert opinions [3, 7, 13]. Corticosteroids are considered to be the mainstay of treatment and other immunosuppressive agents, such as methotrexate, azathioprine, and mycophenolate mofetil, are often used with steroids in patients unable to tolerate high-dose corticosteroids or with a recurrent course. Infliximab, a TNF-α inhibitor, is considered to be the most promising adjunct treatment to steroids for patients with severe disease [14].
The studies of neurosarcoidosis in China are sparse, and most of them are single case reports. In this study, we performed a retrospective analysis to demonstrate the clinical characteristics, diagnosis, and treatment of neurosarcoidosis in eight Chinese patients.
Methods
This study received ethical approval from the Xuan Wu Hospital institutional review board. We retrieved the data of 137 patients diagnosed with sarcoidosis who were consecutively admitted to Xuan Wu Hospital of Capital Medical University from 2000 to 2015. The medical records of cases suspected of or proven to be neurosarcoidosis were reviewed.
The diagnosis of neurosarcoidosis was classified as definite, probable, and possible according to the diagnostic criteria first proposed by Zajicek et al. [15] and modified by Marangoni et al. [16] and Carlson et al. [17]: definite, clinical presentation suggestive of neurosarcoidosis with exclusion of other possible diagnoses and the presence of positive nervous system histology; probable, clinical syndrome suggestive of neurosarcoidosis with laboratory support for central nervous system inflammation (elevated levels of CSF protein and/or cells, the presence of oligoclonal bands, and/or MRI evidence compatible with neurosarcoidosis) and exclusion of alternative diagnoses together with evidence for systemic sarcoidosis (positive histology and/or at least two indirect indicators from FDG-PET, chest CT, and serum ACE); and possible, clinical presentation suggestive of neurosarcoidosis with exclusion of alternative diagnoses where the above criteria are not met. Only patients who met the criteria for definite or probable neurosarcoidosis were included in this study.
The clinical data of recruited patients were then retrospectively analyzed, including demographic information, past medical history, clinical presentation, auxiliary examinations (serum ACE levels, chest CT, CSF assays, MRIs, electromyography, and histology), treatment, and outcome. The lag time between onset of symptoms and diagnosis, history of sarcoidosis, and detailed neurological features were particularly considered.
Results
Eight patients who met the criteria for definite or probable neurosarcoidosis were included (Table 1), accounting for 5.84% (8/137) of all cases with sarcoidosis (Table 2). Six patients (75%) were female. The mean age at onset of symptoms was 50.25 ± 12.07 years (range 33–71), and the mean age at diagnosis was 50.88 ± 11.66 years (range 33–71). The mean lag time between onset of symptoms and diagnosis was 8.19 ± 11.70 months (range 0.5–36). Of those eight patients, five (62.5%) had a prior history of extraneurologic sarcoidosis (Table S1) leading to earlier diagnosis (3.4 vs. 16.2 months).
One patient was diagnosed as definite neurosarcoidosis confirmed by brain and cervical lymph node biopsy (case 1). The other seven cases were classified as probable neurosarcoidosis together with evidence for systemic sarcoidosis, supported by mediastinal lymph node biopsy in five patients (cases 2, 3, 4, 7, and 8), and cervical lymph node biopsy in one patient (case 6) and two indirect indicators in one patient (case 5).
Clinical manifestation
Neurological symptoms and signs were the first clinical feature of sarcoidosis in three cases (37.5%; cases 3, 5, and 8), who were found to have evidence of extraneurologic sarcoidosis on further investigation. No patients presented only neurologic sign as manifestations of the disease. Pulmonary sarcoidosis, found in all patients, was most common type of extraneurologic sarcoidosis. Peripheral lymph nodes were involved in three patients (cases 1, 5, and 6), spleen in two patients (cases 3 and 7), hepatic portal and retroperitoneal lymph node in one (case 7), and skin in one patient (case 3).
Neurological symptoms of eight patients were shown in Fig. 1. The most common symptom was sensory disturbance occurring in six patients, including limb numbness, girdle-band sensation, formication, and neuralgia. Fever, headache, limb weakness, and sphincter disturbance were also common in this group. Disturbance of consciousness and seizures were found only in two patients who were eventually dead (cases 1 and 6).

Neurological symptoms of eight patients with neurosarcoidosis
Auxiliary examination
Serum ACE level was assayed in five patients (Table 1), and it was elevated in only one patient (case 6). Blood calcium levels were normal in all patients. CSF examination was performed in all patients and there was no specific change (Table 1). Elevated levels of CSF protein were found in six patients (cases 1, 3, 4, 5, 6, and 8), and the levels of CSF glucose were slightly reduced in two patients (cases 1 and 7), and CSF oligoclonal band was positive in two patients (cases 5 and 6). Intracranial pressure was notably increased in three patients (cases 1, 3, and 6), of which two patients with pleocytosis and obviously high protein levels eventually died (cases 1 and 6).
Magnetic resonance imaging (MRI) was performed in all patients (Table 1). Meningeal enhancement could be found in four patients (cases 1, 3, 5, and 6). T1 hypointense, T2 hyperintense, and T1 contrast-enhancing parenchymal lesions occurred in gray matter in four patients (cases 1, 2, 3, and 6) and white matter in one patient (case 3). Among them, one patient (case 6) had intracranial lesions with nodular enhancement and mass effect mimicking neoplasms (Fig. 2). In addition, posteriorly located enhancing lesions extending from T2 to T5 in the thoracic spinal cord (Fig. 3) were found in one patient (case 4), and bilateral symmetrical enhancement of Meckel’s cave and the trigeminal ganglions (Fig. 4) were found in one patient (case 5).

Intracranial lesions mimicking neoplasms in neurosarcoidosis (case 6). Axial T1 images (a, d) showed hypointense signals located in the right frontal operculum and parietal lobe, respectively. Axial (b, e) and coronal (c, f) T1 post-contrast images revealed two nodular enhancing lesions with mass effect mimicking neoplasms

Spinal cord lesions in neurosarcoidosis (case 4). Sagittal T2 image (a) showed the hyperintense signals in the thoracic spinal cord. Sagittal T1 post-contrast image (b) revealed posteriorly located enhancing lesions extending from T2 to T5

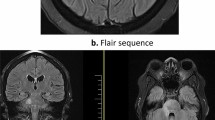
Bilateral trigeminal nerve involvement in neurosarcoidosis (case 5). Compared to the T1 (a, d) and T2 (b, e) images, axial and coronal T1-weighted post-contrast (c, f) MRI showed intense bilateral symmetrical enhancement of Meckel’s cave and the trigeminal ganglions
EMG was performed in three patients, of which two had axonal motor and sensory neuropathy (cases 7 and 8). Chest CT scan was performed in all patients, showing bilateral hilar lymphadenopathy (cases 1–8) and/or pulmonary parenchymal lesions (cases 3 and 5–8). FDG-PET scan was performed in one patient, revealing hypermetabolic activity in mediastinal and cervical lymph nodes (case 5).
Treatment and outcome
All patients were treated with corticosteroids (Table 1). The initial therapy was intravenous methylprednisolone 0.5–1 g for 3–5 days in five patients (cases 4, 5, 6, 7, and 8), or intravenous dexamethasone 10 mg for 10 days or more (cases 1 and 3), or oral prednisone 60 mg/day for 4 weeks (case 2), followed by tapering oral prednisone. One patient with a recurrent course was given azathioprine in addition to corticosteroids (case 3).
The average time of follow-up was 15.13 ± 9.29 months after diagnosis (range 0.5 to 24 months). Two patients got complete remission (cases 4 and 5), three patients had partial improvement (cases 2, 3, and 7), and one patient was stabilized (case 8). Two patients deteriorated and died 2.5 months (case 1) and 1 month (case 6) after the onset of neurological symptoms.
Discussion
This study revealed that the prevalence of clinical neurosarcoidosis in our sarcoidosis series was 5.84% (8/137), similar to that reported in other case series [8, 18, 19]. Neurosarcoidosis was more common in women in our study and the female-to-male ratio was 3 to 1, slightly higher than the previous findings [3, 8, 12, 15, 17, 20]. The mean age at onset was 50.25 years old, later than that reported in some previous studies (range from 33 to 48 years old) [3, 8, 11, 12, 15, 17, 20]. However, an epidemiological study in Japan showed there was a second peak of incidence in the 50–60-year-old females with sarcoidosis [5]. In our study, women were obviously older than men at onset of symptoms (55.5 vs. 34.5 years). This sex-related difference seemed to be one of the reasons for the relatively later age at onset.
Neurological involvement was the first manifestation of sarcoidosis in 37.5% of cases, and none of the patients had isolated nervous system manifestation in this study. This is different from previous studies, which reported that the rate of neurological involvement as the first manifestation was up to 50–70% [8, 11, 12, 15, 20, 21], and as the only feature of the disease was 10–15% [8, 12]. A retrospective case series of 305 patients reported recently showed that the rate of isolated neurosarcoidosis has gone up to 38% [17]. The rate of this study was far lower than those reported in Caucasian populations. This inconsistency may be caused by the difference of prevalence and clinical features of neurosarcoidosis among different areas and races. However, another important reason is that diagnosis is much more challenging when the neurological involvement is the first or only manifestation of sarcoidosis. A tissue biopsy, especially from the nervous system, is needed for a definitive diagnosis. Neurological manifestations of the patients in this study were various, and the most common symptom was sensory disturbance, which was caused by lesions of the thalamus, the spinal cord, trigeminal nerve, and the peripheral nerve. This was consistent with the previous reports, suggesting that the most common symptoms were sensory disturbance (29–46%) and headache (32–37%) [11, 21]. However, headache may be more common (up to 90%) in patients with isolated neurosarcoidosis [12].
CSF changes in this series were similar to the previous results. Typical CSF findings are elevated protein and/or lymphocytic pleocytosis, which are found in approximately 2/3 of patients [7, 11, 17, 21]. CSF oligoclonal band is also reported in 30–50% of patients [3, 11]. Otherwise, reduced CSF glucose, often seen in neoplastic or infectious meningitis, is found in approximately 10–20% of patients with neurosarcoidosis [3, 22]. MRI features in this series were also consistent with the previous findings. Meningeal enhancement is reported to be the most common finding on brain MRI, seen in 30–40% of patients with neurosarcoidosis [3]. The involvement of brain parenchyma on MRI manifests as two types [23]. One is the granulomas with nodular enhancement and mass effect mimicking neoplasms and infectious lesions (Fig. 2). The other presentation is the peri-ventricle and subcortical white matter lesions with or without enhancement mimicking multiple sclerosis. The spinal involvement on MRI usually manifests as intramedullary lesions located in cervical or upper thoracic spinal cord [24] and often extending to three or more spinal segments [13, 25].
Neurological manifestations and investigations revealed that both central and peripheral nervous system were involved in this series, including brain parenchyma, meninges, spinal cord, cranial nerves, and peripheral nerves. Previous studies showed that cranial neuropathy, seen in 50–70% of patients with neurosarcoidosis, was the most common type of neurological damage [3, 7, 11, 13, 24, 26, 27]. Though any cranial nerve can be involved, facial and optic nerves are most frequently affected [7, 11, 13, 24, 26,27,28]. Bilateral peripheral facial palsy, which also occurred in our study, is considered to be one of the significant characteristics of neurosarcoidosis [26]. However, the most common neurological damages in this group were brain parenchymal lesions and meningeal disease, the frequencies of which were considered to be lower than that of cranial neuropathy, approximately 50% and 20%, respectively [10, 27]. This study also reported patients with myelopathy and peripheral neuropathy, which are less common, accounting for approximately 5–15% of patients with neurosarcoidosis [27]. Usually, central nervous system involvement predicts for a worse disease course compared with peripheral nervous system involvement [8, 22, 29]. Our study revealed that disturbance of consciousness, seizures, hydrocephalus, and abnormal CSF assays (increased intracranial pressure with pleocytosis and obviously high protein levels), occurring only in two patients who eventually died, were associated with poor prognosis.
The evidence for systemic sarcoidosis includes positive histology and/or at least two indirect indicators. Marangoni et al. modified the diagnostic criteria for neurosarcoidosis first proposed by Zajicek et al., suggesting that the serum ACE level had a low sensitivity for the diagnosis [16]. Elevated serum ACE level was found in only one of five patients in our study, supporting this opinion. 18F-FDG PET can detect pulmonary and extrapulmonary lesions as similarly or even more accurately than gallium scintigraphy scan [30]. Therefore, FDG-PET is recommended to the patients to look for evidence of involvement outside the nervous system and identify a target for biopsy [26, 31, 32].
Recently, a meta-analysis based on the articles over the last 35 years including 1088 cases showed that about 2/3 of patients with neurosarcoidosis improved with therapy and that the mortality rate among patients was 5% [11]. There are rare reports on the prognosis of neurosarcoidosis among Chinese. Sarcoidosis appeared to be the more benign courses among Chinese and the outcomes are generally good [6, 33]. However, the patients with extrapulmonary manifestations are more likely to relapse and might respond poorly to standard drug therapy [33, 34]. Our study showed that five patients had complete or partial improvement and the remission rate was 62.5%, consistent with the meta-analysis results. However, two patients deteriorated and died in our study, suggesting a poor outcome for neurosarcoidosis among Chinese. Both patients had a history of pulmonary sarcoidosis and none of them took any the nervous system examinations since diagnosis of pulmonary sarcoidosis was made. Lack of careful follow-up and treatment monitoring may be related to relapse and result in extrapulmonary sarcoidosis. This may be one of the reasons for poor outcome results. Another important reason is that none of the patients received alternative immunosuppressive therapy, such as infliximab or cyclophosphamide treatment. Recently, corticosteroids plus alternative immunosuppressive therapy is recommended to be used early on the high-risk patients (hydrocephalus, seizures, encephalopathy, etc.) to obtain a favorable clinical outcome [35, 36]. The meta-analysis also showed that 24% of patients who used corticosteroids alone were switched to second- or third-line therapy [11]. In this series, corticosteroids remained the first-line treatment and only one patient was given azathioprine as well because of the recurrent disease course. Compared with these findings, our results suggested that the application of other immunosuppressants with corticosteroids for neurosarcoidosis still needs to be vigorously advanced in China.
Neurosarcoidosis is difficult to diagnose at the early stage and might be associated with a poor prognosis in China. Therefore, detailed investigations, such as tissue biopsy, if necessary, are recommended for the suspected patients to obtain a definitive diagnosis as early as possible. Alternative immunosuppressive therapy plus corticosteroids should be initiated early in patients with severe involvement of the central nervous system to improve clinical outcome.
References
Iannuzzi MC, Rybicki BA, Teirstein AS (2007) Sarcoidosis. N Engl J Med 357:2153–2165
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Muller-Quernheim J (2014) Sarcoidosis. Lancet 383:1155–1167
Hebel R, Dubaniewicz-Wybieralska M, Dubaniewicz A (2015) Overview of neurosarcoidosis: recent advances. J Neurol 262:258–267
Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC (1997) Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol 145:234–241
Morimoto T, Azuma A, Abe S, Usuki J, Kudoh S, Sugisaki K, Oritsu M, Nukiwa T (2008) Epidemiology of sarcoidosis in Japan. Eur Respir J 31:372–379
Anantham D, Ong SJ, Chuah KL, Fook-Chong S, Hsu A, Eng P (2007) Sarcoidosis in Singapore: epidemiology, clinical presentation and ethnic differences. Respirology 12:355–360
Hoitsma E, Faber CG, Drent M, Sharma OP (2004) Neurosarcoidosis: a clinical dilemma. Lancet Neurol 3:397–407
Gascon-Bayarri J, Mana J, Martinez-Yelamos S, Murillo O, Rene R, Rubio F (2011) Neurosarcoidosis: report of 30 cases and a literature survey. Eur J Intern Med 22:e125–e132
Joseph FG, Scolding NJ (2009) Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry 80:297–304
Stern BJ (2004) Neurological complications of sarcoidosis. Curr Opin Neurol 17:311–316
Fritz D, van de Beek D, Brouwer MC (2016) Clinical features, treatment and outcome in neurosarcoidosis: systematic review and meta-analysis. BMC Neurol 16:220
Nozaki K, Scott TF, Sohn M, Judson MA (2012) Isolated neurosarcoidosis: case series in 2 sarcoidosis centers. Neurologist 18:373–377
Nozaki K, Judson MA (2013) Neurosarcoidosis. Curr Treat Options Neurol 15:492–504
Tavee JO, Stern BJ (2014) Neurosarcoidosis. Continuum (Minneap Minn) 20:545–559
Zajicek JP, Scolding NJ, Foster O, Rovaris M, Evanson J, Moseley IF, Scadding JW, Thompson EJ, Chamoun V, Miller DH, McDonald WI, Mitchell D (1999) Central nervous system sarcoidosis--diagnosis and management. QJM 92:103–117
Marangoni S, Argentiero V, Tavolato B (2006) Neurosarcoidosis. Clinical description of 7 cases with a proposal for a new diagnostic strategy. J Neurol 253:488–495
Carlson ML, White JR Jr, Espahbodi M, Haynes DS, Driscoll CL, Aksamit AJ, Pawate S, Lane JI, Link MJ (2015) Cranial base manifestations of neurosarcoidosis: a review of 305 patients. Otol Neurotol 36:156–166
Yu S, Chen B, Wang J (1998) Nervous system damage in patients with sarcoidosis. Zhonghua Jie He He Hu Xi Za Zhi 21:367–369
Stern BJ, Krumholz A, Johns C, Scott P, Nissim J (1985) Sarcoidosis and its neurological manifestations. Arch Neurol 42:909–917
Pawate S, Moses H, Sriram S (2009) Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM 102:449–460
Leonhard SE, Fritz D, Eftimov F, van der Kooi AJ, van de Beek D, Brouwer MC (2016) Neurosarcoidosis in a tertiary referral center: a cross-sectional cohort study. Medicine (Baltimore) 95:e3277
Joseph FG, Scolding NJ (2007) Sarcoidosis of the nervous system. Pract Neurol 7:234–244
Bathla G, Singh AK, Policeni B, Agarwal A, Case B (2016) Imaging of neurosarcoidosis: common, uncommon, and rare. Clin Radiol 71:96–106
Schwendimann RN, Harris MK, Elliott DG, Menon U, Gonzalez-Toledo E, Zivadinov R, Pressly TA, Kelley RE, Hoque R, Fowler M, Maghzi AH, Etemadifar M, Saadatnia M, Minagar A (2013) Neurosarcoidosis: clinical features, diagnosis, and management. Am J Ther 20:292–299
Sohn M, Culver DA, Judson MA, Scott TF, Tavee J, Nozaki K (2014) Spinal cord neurosarcoidosis. Am J Med Sci 347:195–198
Ibitoye RT, Wilkins A, Scolding NJ (2017) Neurosarcoidosis: a clinical approach to diagnosis and management. J Neurol 264:1023–1028
Terushkin V, Stern BJ, Judson MA, Hagiwara M, Pramanik B, Sanchez M, Prystowsky S (2010) Neurosarcoidosis: presentations and management. Neurologist 16:2–15
Rosini F, Bennett D, Cerase A, Volterrani L, Federico A, Rottoli P, Rufa A (2017) Cavernous sinus syndrome due to neurosarcoidosis in adolescence: a diagnosis not to be missed. Neurol Sci 38:517–519
Ferriby D, de Seze J, Stojkovic T, Hachulla E, Wallaert B, Destee A, Hatron PY, Vermersch P (2001) Long-term follow-up of neurosarcoidosis. Neurology 57:927–929
Nishiyama Y, Yamamoto Y, Fukunaga K, Takinami H, Iwado Y, Satoh K, Ohkawa M (2006) Comparative evaluation of 18F-FDG PET and 67Ga scintigraphy in patients with sarcoidosis. J Nucl Med 47:1571–1576
Kana V, Petersen JA, Ikenberg K, Chappaz A, Gerth-Kahlert C, Appenzeller P, Linnebank M (2016) Teaching NeuroImages: recurrent oculomotor palsies caused by neurosarcoidosis. Neurology 87:e31–e32
Wegener S, Linnebank M, Martin R, Valavanis A, Weller M (2015) Clinically isolated neurosarcoidosis: a recommended diagnostic path. Eur Neurol 73:71–77
Ding K, Xu ZJ, Huang H, Lu WX, Luo WC (2007) Analysis of the treatment and prognosis of 59 patients with sarcoidosis. Zhonghua Nei Ke Za Zhi 46:52–55
Hsieh CW, Chen DY, Lan JL (2006) Late-onset and rare far-advanced pulmonary involvement in patients with sarcoidosis in Taiwan. J Formos Med Assoc 105:269–276
Scott TF, Yandora K, Valeri A, Chieffe C, Schramke C (2007) Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol 64:691–696
O'Connell K, Williams L, Jones J, McCabe DJ, Murphy D, Killeen R, Tubridy N, O'Riordan S, McGuigan C (2017) Neurosarcoidosis: clinical presentations and changing treatment patterns in an Irish Caucasian population. Ir J Med Sci 186:759–766. https://doi.org/10.1007/s11845-016-1539-y
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No. 81100797) and Beijing High-Level Health Talents Training Project (No. 2015-3-068).
Funding
This work was supported by the grants from the National Natural Science Foundation of China (No. 81100797) and Beijing High-Level Health Talents Training Project (No. 2015–3-068).
Author information
Authors and Affiliations
Contributions
FW, DG, and JJ contributed to the study design, analysis, and interpretation of the data. FW, ZL, AZ, and CW contributed to the acquisition of data. FW contributed to drafting the manuscript for content. All the authors (FW, DG, ZL, AZ, CW, and JJ) contributed to revising the manuscript. JJ and FW conducted the study coordination. All the authors gave final approval of the submitted version.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical or institutional review board approval
This retrospective study received ethical approval from the Xuan Wu Hospital institutional review board. Formal consent is not required for this type of study.
Electronic supplementary material
Table S1
(PDF 56 kb)
Rights and permissions
About this article

Cite this article
Wang, F., Guo, D., Liu, Z. et al. Neurosarcoidosis: clinical characteristics, diagnosis, and treatment in eight Chinese patients. Neurol Sci 39, 1725–1733 (2018). https://doi.org/10.1007/s10072-018-3491-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-018-3491-2