
Abstract
Appropriate description may lead to adequate diagnostic and therapeutic measures, and therefore, a simple scheme to categorize and term the imaging findings of acute myelopathy is suggested based on current literature. Assigning imaging findings to five groups, that is (a) “segmental with rash,” (b) “poliolike,” (c) “granulomatous-nodular,” (d) “longitudinally extensive transverse myelitis,” (e) “short-segment ovoid or peripherally located,” provides a rationale to lessen differential diagnoses. The key for understanding, proper description and differential diagnosis is the correlation of two time points: When did the first symptoms appear and when did imaging take place? Early infarction within the first 24 h will show neither swelling nor enhancement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2002 the “Transverse Myelitis Consortium Working Group” [1] tried to unify preexisting diagnostic criteria for acute transverse myelitis (ATM) and confined the time to reach maximum deficits to 3 weeks following the onset of symptoms. Certain chronic inflammatory diseases, for example tuberculosis, sarcoidosis, HTLV-1 have to be ruled out, and all noninflammatory processes whose evolution typically will take more than 21 days are excluded by this definition as well. This proposal has been widely recognised [2–4].
Hence, several metabolic, toxic, paraneoplastic or hereditary causes of myelopathy with a more insidious course and often symmetric extended lesions especially confined to dorsal or lateral columns [3, 4], intramedullary neoplasms and late sequelae of radiation [4] are disregarded in the following.
To exclude spinal infarction [1], the lower time limit for the nadir of maximum deficits allowing the diagnosis of transverse myelitis was given with 4 h. This assumption was confirmed by an analysis of 57 consecutive patients with spinal infarction [5]: The median time for the interval from symptom onset to nadir was 1 h, but the mean, given with 7.8 h (SD ± 23) disclosed wide dispersion.
However, the first step diagnosing symptoms of acute myelopathy must be the urgent exclusion of extradural spinal cord compression.
The underlying etiologies of acute myelopathies cover a broad spectrum, and demyelinating diseases are an important part of them. The discovery of aquaporin-4 antibodies (AQP4-Ab) in 2004, the more precise description of neuromyelitis optica (NMO; 6, 7) subsequently and the clear distinction from multiple sclerosis (MS) were major contributions to the understanding of acute myelopathy, ultimately leading to more targeted therapeutic measures [8]. Interestingly, the mere distinction of complete versus partial ATM [2] may refer to different disease entities, the latter being more often than the first clinical presentation of MS. Certain clinical findings (e.g. rapid progression, spinal shock; 1) seem to predict a dubious prognosis almost regardless of etiology.
Similar to extradural space occupying lesions, also subdural empyema may cause myelopathy. It is characterised by thin, rim-enhancing fluid collections on MRI [9]. Likewise, the key characteristics of spinal vascular malformations and dural arteriovenous fistulae, engorged vessels and flow voids must not be overlooked [10].
Thus, the following is focussed solely on rare diseases of the spinal cord which present acutely, but differ in pathogenesis. To describe relevant details, contrast enhanced series are a prerequisite and additional brain imaging is mandatory in all nonischemic etiologies. The final diagnosis in MS, NMO, and acute disseminated encephalomyelitis (ADEM) is based on presence or absence of certain brain imaging findings [3, 4, 6, 8, 11] and may be contributing in some rare infections as well [12]. Moreover, if an early examination has led to normal or equivocal results one should bear in mind, that it takes several hours for all the lesions discussed below to become evident on structural imaging, even when severe neurological deficits are present [13]. Without diffusion weighted imaging (DWI), only one out of five spinal cord infarcts scanned within the first 15 h after onset could be delineated on MRI [14]. Therefore repeated examination will yield a more precise analysis of the diagnostic problem sometimes. For practical reasons, imaging features are divided in five subtypes (see Fig. 1a).

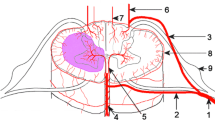
a Imaging features in acute inflammatory processes of the spinal cord. Green: “segmental, with rash”; blue: “polio-like”; orange: “granulomatous, nodular”; pink: longitudinal extensive transverse myelitis; yellow: “short segment ovoid or peripherally located”. b Spinal cord infarcts and presumed vascular territories. a: Spinal sulcal (syn.: sulcocommissural) artery territory infarct; b: anterior spinal artery territory infarct; c: posterior spinal artery/posterolateral spinal artery territory infarct; d: watershed infarcts at the level of the anterior horns between intrinsic and extrinsic systems; e: complete transverse infarct (syndrome of the artery of Adamkiewicz). 1: radiculomedullar artery; 2, 3: posterior and anterior radicular artery; 4: anterior spinal artery; 5: spinal sulcal (syn.: sulcocommissural) artery; 6, 7: posterolateral and posterior spinal artery; 8: vasocorona
Imaging and Clinical Findings
-
a)
“Segmental, with rash”
This lesion type is confined to the level of one or few dorsal roots. Following childhood infection, the virus genome of the varizella zoster virus (VCV) persists in the dorsal root ganglia and the trigeminal and geniculate ganglia as well [4, 12]. In later life VCV may cause shingles, possibly complicated by meningoradiculitis. MRI may show hyperintense lesions on T2 WI and enhancement on post contrast (pc) T1 WI at the dorsal nerve root and their entry zone in the spinal cord pronounced at this level [4]. Rarely, a Brown–Séquard syndrome ensues, representing a hemispinal cord lesion [15].
Quite similar, an ischemic partial transverse syndrome may result from the occlusion of a sulcocommissural artery. But in the syndrome of the sulcocommissural artery consisting of an ipsilateral paresis and crossed dissociated sensation deficit the dorsal column is spared (see Fig. 1b). Six out of 55 patients suffering from spinal cord infarction disclosed this lesion type, all located cervically [16]. Clues for differentiation are the preceding rash in the former and the rapid onset in the latter. In ischemic lesions edema should be absent for at least 24 h after onset.
-
b)
“Poliolike”
Besides the nearly eradicated but prototypical poliovirus, there are other picorna- and some flaviviridiae with similar neurotropism leading to flaccid pareses [3, 4]. As a rule, patients are systemically ill and hence infection cannot be mixed up with the abrupt onset of an acute spinal infarction. MR imaging may point to just a few causative agents for these myelitides, narrowing differential diagnosis, what has been demonstrated again in a recent outbreak of the Enterovirus D 68 belonging to the picornaviridiae family. Imaging mirrors flaccid paralysis and cranial nerve dysfunction [17]: First, T2 WI exhibit hyperintense lesions of the gray matter of the spinal cord (and brainstem) possibly extending lengthwise with some swelling, but in contrast to longitudinally extensive transverse myelitis (LETM; see below; d) sparing adjacent white matter (see Fig. 1a). Within a few days contrast enhancement (CE) of ventral roots on pc T1 WI and hyperintense lesions on T2 WI bordered to the anterior horns appear.
In spinal cord ischemia, similar lesion pattern may occur as so-called “snake–” or “owl’s–eye configuration” (see Fig. 1b). This was observed in 3 out of 55 patients cervically [16], corresponding to a “man-in-the-barrel syndrome”. It is defined by bilateral flaccid proximal paresis of the upper limbs without sensory or motor deficits of the legs.
Exceedingly rare poliomyelitis-like pareses may occur suddenly in one or more limbs days after an asthma attack [18] associated with quite similar lesions. The pathogenesis is still under debate. Interestingly, comparable T2 signal changes within the anterior horns were observed in the beginning of NMO relapses [19].
-
c)
“Granulomatous, nodular”
Although in most cases there is no histological proof, the heading is chosen because nodular enhancements which may be observed in the meninges, the spinal roots or in the spinal cord itself are known to be associated with some parasitic infections, tuberculosis, lues and sarcoidosis, but neoplastic seeding as well (see Fig. 1a) [4, 9, 12]. The nodules may reach from minute and numerous to one or few space-occupying findings. If they accompany longitudinal extensive hyperintense signal changes on T2 WI (see Sect. d), they seem to be more specific than homogeneous enhancements: Likewise, concomitant meningeal findings may disprove the assumed demyelinating nature of an intramedullary enhancing lesion as depicted in Sect. e. True granuloma, for example, sarcoidosis, may exhibit hypointense signal on T2 WI, possibly surrounded by edema.
To prevent diagnostic delay and spinal biopsy, an early screening for systemic manifestation of the underlying disease should be performed [4, 9]. Whereas radicular enhancement on pc T1 WI is typical in lyme borreliosis, nodular intramedullar lesions are exceedingly rare [12].
-
d)
“Longitudinally extensive transverse myelitis (LETM)”
This term was chosen by Wingerchuk et al. [6] to characterize an imaging finding which clearly differentiates NMO (Devic’s disease) from MS and constitutes an important diagnostic criterion for NMO. Both diseases have a predilection for the optic nerves and the spinal cord, but a spinal lesion covering ≥ 3 vertebral segments (defining LETM) is rare in MS [8]. The clinical presentation one would expect is complete ATM [2], but obviously, the pathogenesis of LETM and thereby, deficits and prognosis vary. Even the absence of neurological deficits [12] and excellent recovery in young patients occur despite holocord extension in some cases [20].
The spectrum of etiologies underlying LETM was elucidated by Kitely et al. [21], evaluating clinical and paraclinical data of 76 adult patients with and without the NMO-specific AQP4-Ab. Typical NMO represented with 65 % a majority (49/76, including 5 patients negative for AQP4-Ab). The remaining 35 % (27 patients) were diagnosed as ADEM (5), ADEM positive for an antibody seen in childhood (6), idiopathic ATM (5), infection with known agents (4), MS (3) and other inflammatory, paraneoplastic and vascular diseases (4). More importantly, in selected cases it seemed possible to differentiate monophasic disease with favorable outcome by consistent antibody testing [22].
In NMO, but in other myelitides as well, the extension of LETM may span more than 10 segments [4, 8, 11, 12, 20] and characteristically appears edematous in acute stages. It involves homogeneously central gray and white matter of the spinal cord, being hypointense on T1 WI, and histology discloses swelling and softening [7]. When present, CE in acute NMO is patchy and extensive [8]. In chronic NMO hypointense lesions on T1 WI indicate necrosis and cavitation [6].
More recently, it was proposed to diagnose and treat AQP4-Ab positive patients presenting with a single or recurrent attacks of optic neuritis, myelitis or brain/brainstem disease as a NMO spectrum disease irrespective of LETM to prevent further disabling attacks. The working group of Wingerchuk [23] described those first episodes with short myelitis lesions (< 3 vertebral segments) in 25 out of 319 patients (14 %; see also Sect. e).
ADEM is usually viewed as a monophasic illness triggered by an “antigenic challenge” (infection or vaccination) more often seen in childhood affecting white and gray matter of the brain and spinal cord [21]. To encompass childhood MS, the ADEM consensus criteria [24] require polysymptomatic cerebral illness including “encephalopathy”. A limited or site-restricted spinal form as proposed by Scott [2] is not provided for by now, but could be the adaequate interpretation in cases, for now named “postinfectious” or “idiopathic”: They share trigger (infection, vaccination) and favorable prognosis.
Unfortunately, there are no reliable radiological findings allowing for further distinction in LETM [11], apart from a more frequent involvement of the conus medullaris in AQP4-Ab-negative patients [21]. More important are concommitant phenomena as described in Sect. c. If present, they may argue against NMO and ADEM.
In a large series of 22 patients with known systemic lupus erythematosus (SLE) and myelitis attacks, LETM was observed most often in a subgroup of patients with monophasic, rapid, and devastating course [25]. The time to nadir was below 24 h in the majority of them and all patients presented with fever and emergent spinal fluid examination resembling bacterial meningitis. Most importantly, symptoms of urinary retention developed in 91 % several days prior to any motor symptoms.
Clinically very similar, a rapid adverse course of myelitis may be provoked by certain systemic infections, supposed to be mediated by superantigens [4]. But, intriguingly, early imaging most likely will lead to normal results [12], as it is seen in early spinal infarction without additional DWI. Thus, swelling as evidenced in a first MRI examination strongly argues for an earlier, possibly subclinical onset and clearly differentiates from peracute lesion, notwithstanding the etiology of LETM. There is a variety of infrequent autoimmune diseases being associated with LETM [3, 4, 11] including primary CNS angitis [21, 26] and in some of these, AQP4-Ab were identified as well [27].
But based on the modalities of onset (rapid vs. slow), early imaging findings (swelling, extension lengthwise, enhancement), sparing of the dorsal columns (i.e., syndrome of anterior spinal artery), proof of vertebral body infarct, and anamnestic hints (e.g., aortic dissection, aortic operation), spinal cord infarcts on the one hand and myelitis on the other should stay distinguishable [16]. Interestingly in an own study all complete transverse infarcts (16 out of 55 patients), which might be misinterpreted as LETM most likely, were located in the lower spinal cord especially in the thoracolumbar region (see Fig. 1a and b) [16]. However, in SLE-associated myelitis negative for AQP4-Ab an additional small infarction detected by DWI was reported [4]. Although DWI is a helpful (but technical demanding) tool in spinal cord infarction [16], to describe the diagnostic (and prognostic) value of DWI in LETM remains as an important task [28, 29].
-
e)
“Short segment ovoid or peripherally located”
These lesions predominantly affect white matter and are located most often in the dorsal or lateral columns. In the acute phase they may appear slightly swollen [12], but there is no surrounding edema and hypointense signal changes on T1 WI are uncommon [8]. They should be restricted to ≤ 2 vertebral segments and more often they are located peripherally, abutting the cord surface with a broad basis (see Fig. 1a). Clinical presentation matches the term “acute partial transverse myelitis” introduced by Scott [2]. But because this turns out to be more often the overture of MS (which is to be diagnosed later only with proof of dissemination in time and further lesions elsewhere) the conception “clinically isolated syndrome” (CIS) came into use. Early in the course of MS, there is a clear preponderance of lesion development cervically and thus, one should not be surprised by another lesion, yet the aspect of slight swelling exclusively signifies the acute one.
The abovementioned short lesion type in NMO spectrum disorders tends to be more centrally located and exhibit T1 hypointensity [23, 26]. To acknowledge this and to name NMO as differential diagnosis seems to be important: Immune-modulating substances nowadays prescribed early in MS are potentially harmful in NMO [8, 23] and thus, antibody testing is advisable (see Fig. 2). The pivotal role of brain imaging regarding differential diagnosis of spinal cord lesions is detailed elsewhere and in several references and omitted therefore [4, 8, 11, 12].

a–f: A 36-year-old woman of Asian descent during a first myelitis attack positive for AQP4-Ab, accompanied by nausea and blurring vision. Sagittal T2 WI (a) showing a hyperintense lesion at the level C2–C4 (arrowhead) with inhomogeneous enhancement paramedian right sided on pc T1 WI (c, d: sag.; e, f: ax., arrow; transverse lines in c indicate level of ax. slices e and f). Note prominent hyperintense signal at the obex region (a, black arrow), possibly indicating brainstem manifestation. B: T1 WI sag. disclosing slight hypointense signal changes at the upper and lower border zones (arrows)
At times subacute cervical spondylotic myelopathy may pose a diagnostic problem [30]. Being associated with long fusiform hyperintense lesions on T2 WI and cord enlargement, 40 out of 56 patients (71 %) were initially diagnosed as inflammatory or neoplastic disease [30]. However, a transverse “pancake-like” enhancement just caudal to the site of maximal stenosis suggests that cord compression is responsible. Also, intramedullary lymphoma may present subacutely and diagnosis could be confused with inflammatory disease due to favorable response to steroids and possibly, positive oligoclonal bands in cerebrospinal fluid analysis. However, persisting enhancement months after therapy should raise the suspicion of an underlying neoplasm [3].
Conflict of Interest
The authors (M. Nichtweiß, S. Weidauer) declare that there is no actual or potential conflict of interest in relation to this article.
References
The Transverse Myelitis Consortium Working Group Members. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. 2002;59:499–505.
Scott TF. Nosology of idiopathic transverse myelitis syndromes. Acta Neurol Scand. 2007;115:371–6.
Jacob A, Weinshenker BG. An approach to the diagnosis of acute transverse myelitis. Semin Neurol. 2008;28:105–20.
Goh C, Desmond PM, Phal PM. MRI in transverse myelitis. J Magn Reson Imaging. 2014;40:1267–79.
Nedeltchev K, Loher TJ, Stepper F, Arnold M, Schroth G, Mattle HP, Sturzenegger M. Long-term outcome of acute spinal cord ischemia syndrome. Stroke. 2004;35:560–5.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15.
Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. 2014;24:83–97.
Tackley G, Kuker W, Palace J. Magnetic resonance imaging in neuromyelitis optica. Mult Scler. 2014;20:1153–64.
Wagner M, Klein JC. Meningeal disorders. In: Hattingen E, Weidauer S, Setzer M, Klein J, Vrionis K, editors. Diseases of the spinal cord—novel imaging, diagnosis and treatment. Heidelberg: Springer; 2015. pp. 271–99.
Krings T. Vascular malformations of the spine and spinal cord. Clin Neuroradiol. 2010;20:5–24.
Kitley JL, Leite MI, George JS, Palace JA. The differential diagnosis of longitudinally extensive transverse myelitis. Mult Scler. 2012;18:271–85.
Nichtweiß M, Hattingen E, Weidauer S. Inflammation of the spinal cord. In: Hattingen E, Weidauer S, Setzer M, Klein J, Vrionis K, editors. Diseases of the spinal cord—novel imaging, diagnosis and treatment. Heidelberg: Springer. 2015. pp. 315–68.
Holland NR. Acute myelopathy with normal imaging. J Child Neurol. 2013;28:648–50.
Alblas CL, Bouvy WH, Lycklama À Nijeholt GJ, Boiten J. Acute spinal-cord ischemia: evolution of MRI findings. J Clin Neurol. 2012;8:218–23.
Hosaka A, Nakamagoe K, Watanabe M, Tamaoka A. Magnetic resonance images of herpes zoster myelitis presenting with Brown-Séquard syndrome. Arch Neurol. 2010;67:506–7.
Weidauer S, Nichtweiß M, Hattingen E, Berkefeld J. Spinal cord ischemia: aetiology, clinical syndromes and imaging features. Neuroradiology. 2015;75:241–57.
Maloney JA, Mirsky DM, Messacar K, Dominguez SR, Schreiner T, Stence NV. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 Enterovirus D68 outbreak. AJNR Am J Neuroradiol. 2015;36:245–50.
Yeung SC, Antonio G, Ip KS. Flaccid paralysis of the limbs after an asthmatic attack. Pediatr Neurol. 2010;42:133–6.
Krampla W, Aboul-Enein F, Jecel J, Lang W, Fertl E, Hruby W, Kristoferitsch W. Spinal cord lesions in patients with neuromyelitis optica: a retrospective long-term MRI follow-up study. Eur Radiol. 2009;2535–43.
Yiu EM, Kornberg AJ, Ryan MM, Coleman LT, Mackay MT. Acute transverse myelitis and acute disseminated encephalomyelitis in childhood: spectrum or separate entities? J Child Neurol. 2009;24:287–96.
Kitley J, Leite MI, Küker W, Quaghebeur G, George J, Waters P, Woodhall M, Vincent A, Palace J. Longitudinally extensive transverse myelitis with and without aquaporin 4 antibodies. JAMA Neurol. 2013;70:1375–81.
Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, Palace J, Vincent A. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–77.
Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, Wingerchuk DM, Shuster EA, Jiao Y, Horta ES, Pittock SJ. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol. 2015;72:81–7.
Marin SE, Callen DJ. The magnetic resonance imaging appearance of monophasic acute disseminated encephalomyelitis: an update post application of the 2007 consensus criteria. Neuroimaging Clin N Am. 2013;23:245–66.
Birnbaum J, Petri M, Thompson R, Izbudak I, Kerr D. Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis Rheum. 2009; 60:3378–87.
Eckstein C, Syc S, Saidha S. Differential diagnosis of longitudinally extensive transverse myelitis in adults. Eur Neurol J. 2011;3:27–39.
Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler. 2012;18:5–10.
Marcel C, Kremer S, Jeantroux J, Blanc F, Dietemann JL, De Sèze J. Diffusion-weighted imaging in noncompressive myelopathies: a 33-patient prospective study. J Neurol. 2010;257:1438–45.
Thurnher MM, Law M. Diffusion-weighted imaging, diffusion-tensor imaging, and fiber tractography of the spinal cord. Magn Reson Imaging Clin N Am. 2009;17:225–44.
Flanagan EP, Krecke KN, Marsh RW, Giannini C, Keegan BM, Weinshenker BG. Specific pattern of gadolineum enhancement in spondylotic myelopathy. Ann Neurol. 2014;76:54–65.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nichtweiß, M., Weidauer, S. Differential Diagnosis of Acute Myelopathies: An Update. Clin Neuroradiol 25 (Suppl 2), 183–187 (2015). https://doi.org/10.1007/s00062-015-0401-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00062-015-0401-3