
Abstract
Autoimmune diseases and autoinflammatory diseases have a number of similar etiopathogenetic and clinical characteristics, including genetic predisposition and recurrent systemic inflammatory flares. The first phase of ADs involves innate immunity: by means of TLRs, autoantigen presentation, B and T cell recruitment and autoantibody synthesis. The second phase involves adaptive immunity, a self-sustaining process in which immune complexes containing nucleic acids and autoantibodies activate self-directed inflammation. The link between autoimmunity and autoinflammation is IL-1ß, which is crucial in connecting the innate immune response due to NLR activation and the adaptive immune responses of T and B cells. In conclusion, although ADs are still considered adaptive immunity-mediated disorders, there is increasing evidence that innate immunity and inflammasomes are also involved. The aim of this review is to highlight the link between the innate and adaptive immune mechanisms involved in autoimmune diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term “autoimmunity” used to be referred to a condition associated with the dysregulation of adaptive immunity alone (Table 1), whereas “autoinflammatory” was initially defined solely as a consequence of dysregulated innate immunity (Table 2) [1,2,3,4,5]. Consequently, the pathogenetic mechanisms of autoimmune diseases (ADs) were considered to be exclusively mediated by B and T lymphocytes whose B and T cell receptors (BCRs and TCRs) recognised specific antigens, started the inflammatory response against autoantigens, and activated B cell-mediated autoantibody production whereas the autoinflammatory diseases were defined as unprovoked episodes of inflammation, without high titre of autoantibodies or antigen-specific T cells [6,7,8,9,10]. This view clearly separated autoinflammation and autoimmunity as distinct immunological diseases. It was believed that in autoinflammation, no pathogenetic role was played by adaptive immunity, autoantibodies or autoreactive lymphocytes [1,2,3,4,5]. More than 10 years ago, McGonagle and McDermott first theorised the existence of a “continuum model” of immunology, in which diseases lie on a spectrum from autoimmune to autoinflammatory, with variable contributions of both the innate and the adaptive immune responses to particular diseases [3].
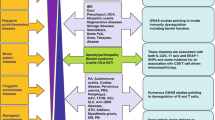
Autoinflammatory diseases are a group of monogenic and polygenic disorders characterised by recurrent inflammatory episodes whose heterogeneous symptoms are frequently associated with fever (Table 2) [1,2,3,4,5]. The key pathogenetic moment in autoinflammatory diseases is the direct and indirect dysregulation of inflammasomes, the multiprotein cytoplasmic complexes characteristic of innate immunity and inflammatory responses [11]. The main components of inflammasomes are members of the nucleotide-binding oligomerisation domain(NOD)-like receptor (NLR) family, a class of pattern recognition receptors (PRR) [11]. When induced to do so by pro-inflammatory triggers such as the stimuli transmitted by pathogens or damaged cells, NLRs respectively detect pathogen-associated or damage-associated molecular patterns ((PAMPs or DAMPs) and begin inflammasome assembly, which generates the proteolytic activation of caspases and the conversion of pro-interleukin(IL)-1β into active IL-1β, a key molecule of inflammation and innate immunity [11].
Although ADs are still considered adaptive immunity-mediated disorders, there is increasing evidence that innate immunity and inflammasomes are also involved [1,2,3,4,5]. It seems clear that despite the differences in the system primarly involved, autoinflammatory and autoimmune disease share common characteristics as the activation against self, with subsequent systemic inflammation, and the absence of an external causal trigger clearly identified. The main difference between the two pathogenetic mechanisms is that in autoinflammatory diseases, the innate immune system is the one directly causing inflammation, while in ADs, it subsequently activates the adaptive system that is the one to activate the inflammatory process.
The link between autoinflammation and autoimmunity: an overview
It is known that innate and adaptive immunity are strictly interconnected. In the case of innate immunity, macrophages, antigen-presenting cells (APCs) and dendritic cells (DCs) act as the first line of host defence [12]. PAMPs and DAMPs are neutralised by NLRs and other classes of PRRs such as Toll-like receptors (TLRs), which recognise various microbial ligands [13], activate inflammasomes, thus increasing the synthesis of pro-inflammatory cytokines including IL1β, IL-18, tumour necrosis factor-alpha (TNF-α) interferon(IFN)-α and IFN-β. These cytokines subsequently activate adaptive immunity [4, 10, 14]. There is increasing evidence that the protracted or increased activation of PRRs plays a key role in autoimmune mechanisms: Leadbetter et al. have shown that the activation of rheumatoid factor-positive B cells mediated by IgG2a/chromatin immune complexes is associated with the synergistic action of the antigen receptor and a protein belonging to the TLR family [15].
This link between TLRs and B cells suggests that TLRs could play a potentially key role in autoantibody responses in various ADs [16,17,18,19,20]. For examples, TLR4 promotes B cell differentiation and leads to the production of antibodies by upregulating the expression of B cell-activating factor (BAFF) [20], a crucial factor that regulates B cell maturation, survival and function, and is upregulated in systemic lupus erythematosus (SLE) [21,22,23].
An important link between autoimmunity and autoinflammation is represented by IL-1ß, which is crucial in connecting the innate immune response due to NLR activation and the adaptive immune responses of T and B cells [24,25,26,27,28,29,30]. It has been demonstrated by Chung et al. that the increased levels of IL-1 following inflammasome activation can induce T cell polarisation (Th17 differentiation) and, synergized with IL-6 and IL-23, is crucial in mantaining cytokine expression in effector Th17 cells [25].
IL-1 has also been implicated in the activation of IFN-γ in memory T cells [26, 27], T cell proliferation (which is also mediated by IL-2 and its receptor) [28], increased B cell proliferation [29], and increased antibody synthesis [30], thus connecting the inflammasome-driven responses to the adaptive immune system in response to exogenous and endogenous signals.
The role of IL-1 has always been considered of crucial importance in autoinflammatory diseases, as demonstrated by the rapid and sustained improvement in disease severity after treatment with IL-1β drug. Other than in monogenic autoinflammatory diseases, IL-1 β has a pivotal role in rheumatoid artrhitis (RA): it has been shown that IL-1 β induces the expression of different proteolytic enzymes, such as metalloproteinases, collagenases and elastases, thus leading to bone erosions and cartilage destruction [11, 31]. However, treatment with IL-1 drug in RA is not as effective as in autoinflammatory diseases, having a modest effect in comparison with other biologic agents blocking TNF pathways [11, 31].
Other evidence of a link between autoimmunity and autoinflammation regard nucleotide-binding oligomerization domain-containing protein 2 (NOD2) [32], a member of a family of pattern recognition molecules with N-terminal Caspase activation and recruitment domains (CARDs) that is also called the nucleotide binding domain and leucine-rich repeat(LRR)-containing family [33, 34]. NOD2 mutations in NACHT domains are responsible for the autoinflammatory granulomatous diseases Blau Syndrome (BS) and early onset sarcoidosis (EOS). At the same time, polymorphisms in LRRs are also susceptibility regions for Crohn’s disease (CD) [33, 34], a disease that is long believed to be on the cross-road between autoinflammation and autoimmunity. In addition, NOD2 is a key molecule recognising bacterial muramyl dipeptide (MDP) [35, 36], the neutralisation of which is abolished in NOD2-deficient mice, thus suggesting that NOD2 may act as an adjuvant receptor for antibody production and play a key role in activating adaptive immunity [32].
Another emblematic example of the link between autoimmunity and autoinflammation is represented by the role of inflammasomes, crucial components of the innate system, in a paradigmatic autoimmune disease such as systemic lupus eritematosus (SLE). Several autoinflammatory disorders, such as CAPS, FCAS, MWS and NOMID, have been linked with mutations in inflammasomes, particularly involving NLRP3 inflammasome, thus leading to increase in release of IL-1 [37]. These mutations have also been linked with polygenic inflammatory disorders, such as gout and pseudogout. In comparison the understanding of the role of inflammasomes in autoimmunity is less clear. In recent years there have been increasing evidence that inflammasomes, particularly NLRP3, could play a potentially key role in SLE, with various mechanisms. Immune complexes formed secondarly to antibody recognition have been shown to stimulate inflammasome activation through upregulation of TLR-dependent activation of NFκB and subsequent activation of the NLRP3 inflammasome [38]. Kidney biopsy findings from SLE patients have shown increased expression of inflammasome components, including NLRP3 and caspase-1 [39]. C3a, which is released during complement activation in tissues in SLE, regulates ATP secretion in inflammasome thus causing their activation and subsquent production of IL-1 [40]. Anti-dsDNA antibodies (Abs) play critical roles in the development and progression of SLE. The activity of caspase-1 was significantly increased in active SLE patients and was correlated with serum levels of anti-dsDNA antibodies and with disease activity. It was observed that anti-dsDNA Abs activated NLRP3 inflammasome in monocytes/macrophages from SLE patients by binding to TLR4 and inducing the production of mitochondrial ROS [41]. Moreover, activation of NLRP3 is involved in the pathogenesis of podocyte injuries and the development of proteinuria in lupus nephritis [42].
Autoinflammatory diseases
Autoinflammatory diseases are a group of monogenic and polygenic disorders characterised by dysregulation in the innate immune system, particularly in key cytokine pathways such as the ones involving TNF and interleukin 1, including the complex of adaptor molecules known as inflammasome, as well as mutations in proteins associated with bacterial sensing. They share as clinical manifestations recurrent episodes of unprovoked inflammation, often accompanied by fever and a wide range of systemic manifestations involving cutanous, vascular and musculoskeletal systems in the absence of autoantibodies or antigen specific T-lymphocytes. The first autoinflammatory disease described, and also the most common, was the mediterranean familial fever, followed by TNF receptor-associated periodic syndrome (TRAPS) in 1999, when the autoinflammatory disease term for these diseases was proposed by McDermott and collegues. Since then an increasing number of diseases were classified, with a wide range of monogenic and polygenic mutations identified in the innate immune pathways [3].
The hereditary periodic fevers are familial Mediterranean fever (FMF), mevalonate kinase deficiency (MKD), and TNF receptor-associated periodic syndrome (TRAPS) [43,44,45]. FMF and MKD are respectively due to Mediterranean fever (MEFV) and mevalonate kinase (MVK) gene mutations codifying pyrin protein and mevalonate kinase (MK) enzyme (MK), and associated with enhanced procaspase-1 activation and increased IL-1β processing and secretion, whereas TRAPS is due to mutations in TNFRSF1A, whose intracellular accumulation in the endoplasmic reticulum (ER) leads to the activation of ER-stress responses and the mitochondrial release of reactive oxygen species (ROS), with the upregulation of pro-inflammatory cytokines such as IL-1β, TNF-α and IL-6 [43, 44]. As seen before, the autoinflammatory granulomatous diseases Blau syndrome (BS) and early-onset sarcoidosis (EOS), are caused by NOD2 mutations in NACHTdomains. NOD2 polymorphisms in LRRs are also susceptibility regions for Crohn’s disease (CD) [33, 34].
The adaptive immune response is also potentially linked to innate immunity through the Th1 and Th17 cell responses mediated by the nucleotide oligomerisation domain-like receptor family, pyrin domain-containing 3/caspase-1 (NLRP3) inflammasome, mutations of which are responsible for the autoinflammatory diseases known as cryopyrinopathies (CAPS) [43, 44].
In addition to hereditary periodic fevers, granulomatous diseases and CAPS, an autoinflammatory group includes pyogenic diseases, adenosine deaminase 2 (ADA2) protein deficiency, and NLRC4-mediated diseases [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. The pyogenic MAIDs include pyogenic arthritis pyoderma gangrenosum and cystic acne syndrome (PAPAS), Majeed syndrome, and interleukin-1 receptor antagonist (DIRA) deficiency, which are respectively due to mutations in PSTPIP1, LPIN2, and IL1RN genes [45,46,47,48]. MAIDs also include diseases mediated by adenosine deaminase 2 (ADA2) protein deficieny, an increased synthesis of type I IFN) proteasome dysfunction, and IL-18 dysregulation [50]. The more recent identification of autosomal-recessive mutations in the CECR1 gene leading to ADA2 protein deficiency has been found to be responsible for the early-onset febrile vasculitis associated with stroke [51, 52]. Other recently described monogenic autoinflammatory diseases are the IFN-mediated autoinflammatory diseases (IMADs) due to genetic mutations leading to an increased synthesis of type I IFN, which include Aicardi–Goutières syndromes (AGS) and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE syndrome) [53,54,55,56,57,58,59]. Another recently identified, rare monogenic autoinflammatory disease directly involving the inflammasome complex and caused by mutations in NLRC4 gene is an autoinflammatory syndrome with enterocolitis that is associated with susceptibility to macrophage activation syndrome (MAS) [60, 61]. Physiologically, NLRC4 is an adapter protein for caspase-1 activation that cooperates with cytosolic receptors for various bacterial proteins (NAIPs). NLRC4/NAIP oligomerisation leads to the recruitment of the adaptor protein apoptosis-associated speck-like protein enclosing a CARD (ASC) [61,62,63,64], which allows the NLRC4 inflammasome to activate pro-caspase-1, thus increasing IL1β synthesis and cell inflammation [61,62,63,64].
The main pathogenetic aspects of monogenic autoinflammatory diseases are shown in Table 2.
Greater understanding of the main pathogenetic mechanisms underlying a number of polygenic disorders has led to them being considered autoinflammatory disorders [1]: these include periodic fever, aphthous stomatitis, pharyngitis and adenitis syndrome (PFAPAS), systemic-onset juvenile idiopathic arthritis (So-JIA), adult-onset Still’s disease (AOSD), macrophage activation syndrome (MAS), Schnitzler syndrome, chronic recurrent multifocal osteomyelitis (CRMO) and synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome, pyoderma gangrenosum, Sweet’s syndrome, and CD [1, 64]. However, more than 10 years ago, McDermott and McGonagle suggested that a number of these diseases, classically considered to be exclusively autoimmune, were on the borderline between autoimmune and autoinflammatory diseases with a prevalent inflammatory component [3].
From autoimmune to autoinflammatory genetics
Genetic variants of the major histocompatibility complex (MHC) or the human leukocyte antigen (HLA) region located on the short arm of chromosome 6 are associated with autoimmune and inflammatory diseases [65, 66]. Fernando et al. made a pooled analysis of different studies investigating the causal links between MHC variants and various autoimmune and inflammatory diseases, and found that HLA-DR2 and HLA-DR3 were alleles inducing susceptibility to SLE and multiple sclerosis (MS) among Europeans, and that several HLA-DR4 haplotypes may predispose to SLE (HLADRB1* 0401) and MS (multiple HLA-DRB1*04 alleles) [65]. HLA-DRB1*0401 and *0405 are associated with RA, whereas haplotypes DR3, DR4 and DR9 are associated with type 1 diabetes (T1D), class I MICA5.1 and HLA-A19 and class II HLA-DR5, -6, -8, and -10 are associated with inflammatory bowel disease (IBD) [65]. Various HLA-DR1 haplotypes have been associated with a predisposition to CD, ulcerative colitis (UC) and RA, and various DR2 haplotypes have been observed in SLE, MS, and UC [65]. HLA-DR3 haplotypes predispose for SLE, MS and T1D, and HLA-DR9 haplotypes predispose for RA and T1D [65]. HLA-DR4 is a shared disease susceptibility allele in RA, SLE, CD, MS, and T1D [65].
Autoimmune diseases with distinctive autoantibody profiles and diseases seronegative for autoantibodies are respectively associated with MHC class II and class I alleles [67,68,69,70], and HLA-DR3-DQ2, HLA-B8 and HLA-A1 alleles are associated with diseases such as celiac disease, T1D and autoimmune thyroid diseases (also due to linkage within the MHC locus) [67,68,69,70,71].
In the case of autoimmunity both mechanisms involved in the binding between self-peptide and the MHC complex and post-translational modifications in self-peptide can be involved in the pathogenesis of autoimmune disease in subjects with an HLA risk allele [45, 67]. For example, a change from arginine to citrulline and the synthesis of autoantibodies against citrullinated peptides and proteins (anti-citrullinated protein antibodies [ACPAs]) can be detected years before the clinical onset of RA, and their identification may be predictive of RA in subjects with HLADRB1* 04:01 and *04:04 [67, 72,73,74,75,76]. Post-translational modification can also be involved in the pathogenesis of autoimmune diseases by affecting the uptake of antigen by APCs [67, 76].
In patients with Behçet’s disease, ankylosing spondylitis (AS) and psoriasis, there is an associations between polymorphisms in the aminopeptidase gene endoplasmic reticulum aminopeptidase 1 (ERAP1) and disease-associated MHC class I alleles [77, 78].
In RA patients, the P4 and P6 pockets of HLA-DR proteins have been identified as the most important polymorphisms associated with susceptibility [79, 80]. Autoimmune diseases with autoantibodies show associations between MHC class II and various adaptive immune genes; in the case of RA, there is an association with cytotoxic T lymphocyte sntigen-4 (CTLA4) and protein tyrosine phosphatase non-receptor type 22 (PTPN22) [81, 82].
There are genetic overlaps between innate immunity-mediated diseases such as CD and some MHCs [83]. For example, an association has been reported between CD and MHC class I-associated diseases that represents a disease at the crossroads between autoinflammation and autoimmunity. Furthermore, despite the association with MHC (mainly MHC class I), it has been found on the basis of recurrent systemic clinical findings and the response to IL-1 inhibition that other polygenic diseases such as So-JIA, and AOSD are linked to predominant innate immunity involvement [81, 84, 85].
The phases linking innate and adaptive immunity provide that peptide/MHC (pMHC) class II tetramers play a predominant role in the interactions between T cells and APCs [79,80,81,82]. Self-peptide/MHC complexes on thymic APCs are essential for developing T cells [83], and MHC class II tetramers are important for characterising CD4 T cell specificity. Selection against high-affinity self-Ag recognition influences the avidity of the interactions between a TCR and its pMHC [86,87,88]. Specific APCs located in the thymic cortex and medulla can be key factors in coordinating the selection of a functional and self-tolerant T cell repertoire, and allowing the activities of mature T cells in the periphery [89].
A number of autoinflammatory mechanisms related to monogenic mutations can be found during the course of various autoimmune diseases [90] including autoimmune lymphoproliferative syndrome (ALPS), which is characterised by adenomegaly, splenomegaly, cytopenia, autoantibodies, and SLE-like characteristics, and caused by mutations in the genes involved in the Fas-mediated apoptosis pathway [90, 91].
Various innate immunity-related genes may help in clarifying the phenotype and pharmacogenomics of autoimmune conditions; for example, the IL-1α 889T allele can be associated with the a lack of response to cyclophosphamide in systemic sclerosis (SSc) [90, 92].
It has been reported that numerous genetic polymorphisms of inflammasomes are associated with susceptibility, disease activity and therapeutic response of a number of autoimmune diseases [86]. These include single nucleotide polymorphisms (SNPs) affecting the priming of inflammasomes, and the TLR7 and TLR9 polymorphisms associated with SLE [93,94,95,96,97]. Furthermore, polymorphisms of the purinergic P2X7 receptor (P2X7R), a key molecule for ATP release from damaged cells that can cause the potassium efflux activating the NLRP3 inflammasome have been associated with susceptibility to RA, MS, SLE and lupus nephritis in different populations. [93, 98]. The increased priming of inflammasomes in autoimmune diseases has also been reported to be associated with pro-inflammatory stimuli, including apoptotic cell debris [24, 39, 93, 99, 100].
NLRP1 and NLRP3 play a key role in tissue damage, and NLRP1 SNPs are associated with SLE, RA, SSc and vitiligo in various populations, and NLRP3 SNPs are associated with So-JIA and increased inflammation in autoimmune diseases [93, 101,102,103,104].
Another link between the innate and adaptive sides of immunity can be found in the occurrence of autoimmune manifestations during the course of primary immune deficiency disorders, which have an early onset and are associated with a family history of severe and atypical autoimmune diseases [105, 106]. A deficiency in complement factors (mainly C1, C4 and C2) leading to alterations in the mechanisms of central tolerance and the clearance of apoptotic cell debris are closely associated with SLE [105]. Impaired central and peripheral tolerance leads to two primary immune deficiency disorders: autoimmunity, polyendocrinopathy, candidiasis, ectodermal dysplasia (APECED) and immune dysregulation, polyendocrinopathy, X-linked syndrome (IPEX) [105, 107,108,109]. APECED is an autosomal recessive disease caused by mutations in the autoimmune-regulator (AIRE) gene, and IPEX is an X-linked syndrome due to mutations in forkhead box P3 (FOXP3), a key regulator of T regulator cells (TRegs) [105, 107,108,109].
Defects in B cell tolerance with a lack of mature B cells and immunoglobulins gives rise to congenital agammaglobulinemia and possible autoimmunity. Autoimmunity can also occur during the course of common variable immune deficiency (CVID), in which there are autoreactive B cells despite the reduction of isotype-switched memory B cells [105, 110,111,112].
Various mechanisms also contribute to autoimmunity in Wiskott–Aldrich syndrome (WAS), which is caused by mutations in the gene encoding WAS protein (WASP), which is essential for cytoskeletal structure: impaired tolerance causes alterations in the negative selection of autoreactive B cells [105, 113, 114].
T cell impairment involving central tolerance may also be closely associated with autoimmunity and, in the case Tregs, both central and peripheral tolerance may be involved [105, 115].
Components of innate and adaptive immunity during the course of autoimmune diseases
DCs are APCs capable of stimulating the clonal expansion of naïve T cells, impairing tolerance of self-reactive lymphocytes, and inducing autoimmune mechanisms in ADs [116,117,118,119,120]. DCs play a significant role in breaking tolerance of self-reactive lymphocytes and in supporting autoimmune responses. Low- molecular-weight chemoattractant proteins recognised as chemokines contribute to recruiting DCs and immune cells (including T and B cells), thus leading to the formation of lymphoid tissue structures within target tissue [116,117,118,119,120]. DCs can also guide naïve T cells to become Th1, Th2 or Treg cells by means of cytokine IFN-γ, IL-4 or IL-10 stimulation [116,117,118,119,120].
As part of the innate immune system, DCs have been identified within cellular infiltrates presents in salivary glands of patients with Sjögren’s syndrome, in the synovium of RA patients and in the lymphoid tissue present in autoimmune thyroid disease, also in the skin lesions identifiable in patients with psoriasis [116].
T and B cell clonal selection is essential for the adaptive immune system and, as T and B cell repertoires are extremely different, cell clones can be divided into naïve and memory subsets [121]. The naïve T cell repertoire contains distinct T cell clones (effector and memory T cells), memory B cells and plasma cells [121]. Memory T cell subsets expand in response to in vivo antigen stimulation. They are functionally widely heterogeneous, and can also differentiate into pathogen-specific memory cells when guided by specific microbial patterns [121,122,123,124]. During immune responses, antigen-specific naïve B cells clonally expand and are selected by means of germinal centre reactions to differentiate into memory B cells [121,122,123,124]. B cells also differentiate into plasma cells, which are capable of synthesising and secreting antibodies, and their persistence in bone marrow leads to steady serum antibody levels [121,122,123,124]. B cells produce immunoglobulins, secrete cytokines, and act as APCs [119]. They also express clonally rearranged, antigen-specific BCRs and TLRs, thus combining both adaptive and innate immunity [119, 120]. By the expression of both BCR and TLR B cells integrate antigen specific and danger associated signals. It was proposed that, based on functional responses and their BCR repertoire, naïve B cells can be divided as innate like or adaptive like cells [125], the first ones able to generate rapid antibody responses independent of T cell stimulation, wether the latter participants in T dependant responses that lead to high affinity antibodies production. Activation of different TLRs on these cells is important to differentiate their immune responses.
Dual BCR and TLR signals can markedly promote the activation of self-reactive B cells by uptaking antibody/DNA complexes via a rheumatoid factor-specific BCR [125, 126]. Variations in TLR expression and signalling can modify B cell responses, and so individual TLR expression profiles allow various effector B cell populations to manifest specific forms of antibody production, cytokine secretion and antigen presentation [125, 127, 128]. Cooperation between TLR, BCR and B cell-intrinsic MYD88 signals plays a key role in B cell responses to different pathogens (mainly viruses), through these characteristics B cells are crucial in linking innate and adaptive immune mechanisms [125, 129].
Impaired tolerance of self-antigens or modified self-antigens due to the non-clearance of apoptotic debris, inflammation-mediated modification of self-antigen, or cross-reactivity between non-self and self-antigens are responsible for the synthesis of autoantibodies. Some autoantibodies can cross-react between an extra- or intracellular ligand TLR and the cell surface, thus leading to the perpetuation of B cell activation and autoantibody-mediated responses [130,131,132,133,134]. The synthesis of (mainly IgG) autoantibodies can lead to inflammation with the release of intracellular or modified self-antigens, thus determining antibody effector function and the proliferation of the autoreactive B cell clones [130]. Given their ability to recognise various self-antigens, TLRs also play a key role in starting autoantibody responses and acting synergistically with Fc-mediated effector functions [130,131,132,133,134,135].
Non-haematopoietic stromal cells, resident leukocytes and infiltrating immune cells control inflammation by supporting immunological defences against infection, trauma, injury and cancer [136]. Their good communication and function lead to host defence, antigen removal, and adequate control of tissue damage, but autoimmunity and chronic and severe disease can occur in the case of the perpetuation or inappropriate regulation of these mechanisms [136].
Ectopic lymphoid-like structures (ELS) are lymphoid aggregates containing lymphocytes, monocytes, macrophages and DCs, and may be simple B and T cell aggregates or well-organised, functional germinal centres [136].They often develop at inflammatory sites and act as an essential part of the immune response by affecting both protection and progress during the course of transplant rejection, autoimmune infections and malignancies [136]. In ADs, the accumulation of autoreactive B cells in ELS maintain autoimmunity against specific antigens and self-antigens [136]. It has been suggested that EBV infection may remain latent and promote B cell survival and proliferation [136]. Ectopic follicles in the organs of subjects with autoimmune diseases such as RA and Sjögren’s syndrome frequently show latent EBV infection, leading to the belief that Epstein–Barr virus (EBV) may also play a role in the development of autoimmunity [136].
Other cells involved in the pathogenesis of autoimmunity are T cells [137]. TCRs are heterodimers consisting of two disulphide-linked chains: one α and one β chain, or one γ and one δ chain [138, 139]. All TCR chains have a constant and a variable domain with six hypervariable loops recognised as complementarity-determining regions (CDRs), a membrane-spanning region, and a cytosolic tract [138, 140]. The TCRs located on T cells are the primary units recognising pMHC [138]. In the thymus, the binding of TCRs and pMHC leads to cell differentiation, and some thymocytes differentiate into mature T cells [138, 141]. In secondary lymphoid organs, the binding provides a complex differentiation programme associated with effector action [138, 142].
In SLE, serum and organ clonal T cell expansion may be reactive to antigens, autoantigens such as nucleosomal histones and U1 small nuclear ribonucleoprotein A, and drive tissue inflammation and damage [137, 143,144,145,146,147,148,149]. Thapa et al. made a longitudinal analysis of peripheral blood T cell receptor diversity in patients with SLE using next-generation sequencing and, in comparison with healthy controls, found a significant decrease in T cell diversity in peripheral blood [137].
Glycolysis, lipid oxidation and mitochondrial processes are also essential in regulating the activation, proliferation, and differentiation of CD4+ T and memory cells [150, 151]. In a recent study, Yin et al. found that impaired T cell metabolism was a key factor in the pathogenesis of SLE and suggested that it might be an appropriate therapeutic target [150]. They also reported that CD4+ T cells from a B6.Sle1.Sle2.Sle3 lupus-prone mouse model showed increased ex vivo and in vitro glycolysis and mitochondrial oxidative metabolism in comparison with non-autoimmune controls [150].
Another T cell population involved in the pathogenesis of autoimmunity is that of Th17 cell, which can express transcription factor RORγτ and synthesise a number of chemokines. The IL17 axis cytokines include IL-17A-IL-17F, IL-22, TNF-α and IL-6, which have pro-inflammatory activity in autoimmune and autoinflammatory diseases [152,153,154,155,156,157,158,159]. Th17 differentiation is due to the cooperative action of TGF-β and other pro-inflammatory cytokines such as IL-6 and IL-23, that is involved in the immune action of Th17 cells at the level of various tissues including skin, lung, and mucosal tissue. The dysregulation of Th17 cells is crucial for autoimmunity and inflammation. Two types of Th17 cells have been identified in vivo, differencing from one another for the property to express IL-10 or IFN-γ. IL-1 stimulation during the polarisation phase, strongly suppresses IL-10 and promotes IFN-γ induction, thus these two kind of Th17 cells, that posses either proinflammatory or anti-inflammatory functions, can be modulated by IL-1β [160]. As seen before, autoinflammatory syndromes are characterised by overproduction of IL-1β caused by dysregulation of inflammasomes. Noster and collegues have demonstrated that in the autoinflammatory disease Schnitzler syndrome, a loss of anti-inflammatory TH17 cells is present, and that physiological levels could be restored after therapeutic IL-1β inhibition. This could have implications for disease pathogenesis and future therapeutic strategies [161].
IL-27 downregulates Th17 as a result of IL-10 synthesis and release [152, 153], whereas the absence of TGF-β, IL-10 and Th-17 cell dysregulation leads to inflammatory responses during host defence inflammation and autoimmune processes [152, 162]. TGF-β and IL-6 can induce a population of IL-17 and IL-10 double-producing cells that are not capable of inducing experimental autoimmune diseases; IL-23 stimulation of TGF-β/IL-6-dependent T cells is necessary for T cells to play a pathogenetic role [163]. IL-6 and IL-23 are regulatory cytokines that promote defensive responses in the phase of interaction between the host and the environment [152, 162].
Tregs are critical in ADs because of their effect on immune homeostasis and the maintenance of peripheral self-tolerance. Their development and function are controlled by the Foxp3 transcription factor [155, 156]. Altered Treg development, function and balance are associated with the development of inflammation and autoimmune mechanisms, whereas the homeostasis of Tregs and effector T cells (Teffs) balance tolerance and effector immune responses [164, 165]. In a study of murine autoimmune arthritis, Chevalier et al. found that Tregs had a stable phenotype and were fully functional in in vitro suppression assays. However, their expansion was late compared with Teffs (T follicular helper cells and Th17 cells) during the early autoimmune phases, and this imbalance is likely to have induced autoimmunity as aresult of insufficient Treg control of Teffs [164]. The dysregulation of thymic and peripheral Treg and Teff homeostasis drives the induction and progression of autoimmunity, and even a short period of insufficient Treg control of autoreactive Teffs may allow the onset of subsequently uncontrolled autoimmunity [164]. The inflammatory milieu generated by disease onset may then further deregulate Treg/Teff homeostasis, and exacerbate the course of the disease course. The authors identified IL-2 and IL-21 as key players in disturbed peripheral Treg and Teff homeostasis as inflammation and lymphopenia triggered autoimmunity by suppressing the expansion of IL-2-controlled regulatory T cells and increasing the expansion of IL-21-mediated effector T cells [164].
Discriminating self and non-self is key element in responding to microbial antigens and inhibiting autoimmunity [166, 167]. Immunological self-tolerance is the absence of responsiveness to self-antigens and is supported by various mechanisms: central tolerance eliminates potentially self-reactive T cells in the thymus, and peripheral tolerance control the T cells that by-pass thymic selection and move to the periphery [166, 168,169,170]. CD4+CD25+ Tregs have an immunosuppressive function and mediate immunological self-tolerance by suppressing the potential suppression of autoreactive T cells [166, 168,169,170], and an altered number and/or function of CD4+CD25+ Tregs has been associated with severe autoimmune diseases, including RA and SLE [166, 171, 172].
The complement system,a key component of the innate immune system, represents another link between innate and adaptive immunity. Its main function is to recognise and eliminate pathogens by means of direct killing or to stimulate phagocytosis in modulating adaptive immunity, thus bridging innate and adaptive responses [173, 174]. When complement mechanisms are unbalanced, the complement system may cause damage by mediating tissue inflammation [173, 175]. Dysregulation of the complement system has been implicated in the pathogenesis and clinical manifestations of a number of autoimmune diseases, including RA, SLE, Sjögren’s syndrome, the vasculitides, dermatomyositis, SSc, and antiphospholipid syndrome [173, 175]. Complement deficiencies have also been associated with an increased risk of developing autoimmune disorders. [173, 175, 176], and various SLE phenotypes may be associated with a congenital deficiency in the early components of the complement system (C1q, C2, and C4) [90, 91]. This makes the complement system a potential therapeutic target by inhibiting complement activation components, complement receptors, and the membrane attack complex [173].
Autoimmune diseases are a direct effect of tissue and organ damage mediated by autoreactive immune mechanisms [177, 178] and, in this context, a key role is playes by autoantibodies, such as rheumatoid factor or antinuclear antibodies, which are totally absent in autoinflammatory diseases [1, 177, 178].
Innate and adaptive immunity as the main therapeutic target
The relationship between innate and adaptive immunity is the main target of immunosuppression, and the relationships between different cell types mediated by soluble molecules such as cytokines, or by cell-to-cell interactions, or by ligand/receptor binding, are therapeutic targets in both ADs and autoinflammatory diseases [179, 180].
Agents interfering with the activation, differentiation or survival of immune cells or indirectly blocking cell interactions or neutralising soluble molecules are all means of suppressing immune responses [179, 180]. Immunosuppression induced by corticosteroids, cytotoxic agents or target-specific monoclonal antibodies has become the mainstay of AD treatment, and has also been reported to be efficacious in autoinflammatory diseases [1, 179,180,181]. In addition to inhibiting the synthesis of prostaglandins, steroids regulate the adaptive and innate immune systems mainly by inhibiting the synthesis of pro-inflammatory cytokines such as IL-2, IL-6 and IFN-γ [179, 181], and other targets of immmunosuppression are components of innate immunity (macrophages, DCs and NK cells) and adaptive immunity (T and B cells) [179, 180].
Anticytokine therapies are particularly successful in the case of ADs with a strong inflammatory component [179]. Blocking TNF-α leads to favourable outcomes in RA, AS, psoriatic arthritis, psoriasis, and inflammatory bowel diseases, and a number of biological agents are available that neutralise TNF-α or block its receptor [179, 182, 183]. They have also been reported to be efficacious in refractory autoinflammatory disorders, such as FMF and TRAPS, particularly in the case of articular involvement [184,185,186].
On the other hand, immunity can be directed against anti-cytokine therapy. In particular, the anti-TNF-α immunogenicity (mainly against the chimeric mouse/human anti-TNF-α IgG1 antibody infliximab [IFX]) due to anti-chimeric antibody (HACA) formation can occur after monotherapy, leading to a loss of efficacy and side effects such as infusion-associated reactions [187, 188]. In order to avoid IFX immunogenicity, it is suggested to use concomitant methotrexate-induced immune suppression [187]; concomitant immunosuppressive therapy also decreases the formation of HACAs [188].
In relation to other anti-cytokine treatments, IL-1 inhibition is useful in various ADs. Anakinra, a recombinant human IL-1 receptor antagonist that competes with IL-1α and IL-1β and thus inhibits the pro-inflammatory effects of both is approved for use in RA at a recommended subcutaneous dose of 100 mg/day. It has been widely used to treat monogenic and polygenic autoinflammatory conditions, and leads to sustained disease remission [189, 190].
Given the effects of IL-6 on the immune system and inflammatory processes, IL-6 antagonism is a potential therapeutic strategy in both autoinflammatory and autoimmune disorders [191,192,193,194,195]. IL-6 is a pleiotropic cytokine secreted by various cell types (T and B cells, macrophages, osteoblasts, fibroblasts, keratinocytes and endothelial cells). is involved in numerous immune pathways, and plays a pivotal role in regulating immune response [193]. Tocilizumab is a humanised monoclonal antibody that specifically inhibits IL-6 by competitively blocking the binding site on the IL-6 receptor, and is approved for the treatment of RA [194].
In addition to TNF, the TNF superfamily (TNFSF) includes a number of the ligand/receptor mechanisms involved in the pathogenesis of autoinflammatory and autoimmune diseases. Its members can initiate immune activation, tissue inflammatory responses, and cell death or suppression, and have been evaluated as possible therapeutic targets in order to control cell responses or restore tolerance [196, 197]. Among these, BAFF is a crucial factor that regulates B cell maturation, survival and function, and is upregulated in SLE [21,22,23]. The BAFF inhibitor belimumab can improve musculoskeletal and mucocutaneous SLE disease activity [198].
Rituximab is a chimeric monoclonal antibody against CD20, a specific B cell differentiation membrane antigen that participates in B cell activation and proliferation [199]. It is administered intravenously and has been approved for use in lymphomas (375 mg/m2 a week for four cycles) [199] and RA (two 1 g infusions separated by an interval of 2 weeks, with repeated courses decided on the basis of an individual clinical evaluation) [200]. The off-label use of rituximab to deplete peripheral CD20-expressing B cells in patients with other immune-mediated diseases has been increasing [201,202,203], but there are no data concerning its use in autoinflammatory diseases.
Other agents targeting IL-17 (sekukinumab) [204] or the p40 chain of IL-12 and IL-23 (ustekinumab) [205], or inhibiting T-cell co-stimulation and activation (abatacept) [206], have been reported to be efficacious in inflammatory arthritides such as RA and PsA [207] but, once again, there are no data concerning its use in autoinflammatory diseases.
Recent studies of therapeutic tolerance in the setting of human transplantation have suggested tolerogenic mechanisms as potential targets [180]. Therapeutic tolerance is the resetting of immune tolerance to autoantigens or the use of tolerance towards alloantigens without the need for immunosuppression [180]. The main cell therapies that have shown promise in human tolerogenesis are haematopoietic chimerism, multipotent mesenchymal stromal cells, the in vivo tolerogenic manipulation of T cells, and the use of exogenous Tregs and DCs [180].
Conclusions
ADs and autoinflammatory diseases have a number of similar etiopathogenetic and clinical characteristics, including genetic predisposition and recurrent systemic inflammatory flares [6,7,8].
The first phase of ADs involves innate immunity: by means of TLRs, DCs recognise and internalise autoantigens arising from the process of apoptosis, which leads to IFN-α production, DC maturation, autoantigen presentation, B and T cell recruitment, and autoantibody synthesis [6,7,8,9, 17]. The second phase involves adaptive immunity, a self-sustaining process in which immune complexes containing nucleic acids and autoantibodies are internalised by DC by means of Fcγ-receptors (FcγRs), thus leading to IFN-α synthesis, additional DC and T cell activation, and autoantibody production [6,7,8,9, 126].
Various data indicate that innate and adaptive immunity represent two strictly interconnected phases in the development of ADs [1,2,3,4,5]. The protracted or increased activation of PRRs plays a key role in autoimmune mechanisms [15], and the strict link between TLRs and B cells suggests that innate immunity may play a key role in inducing autoantibody responses [16,17,18,19,20]. Another important link between autoimmunity and autoinflammation is IL-1ß [25,26,27,28,29,30,31,32, 208], which is crucial in connecting the innate immune response due to NLR activation and the adaptive immune responses of T and B cells [25,26,27,28,29,30,31,32, 208,209,210]. It has been reported that the increase in IL-1 levels following inflammasome activation induces T cell polarisation (Th17 differentiation) [25]. Adaptive immune responses are potentially linked to innate immunity through the Th1 and Th17 cell responses mediated by the NLRP3 inflammasome, mutations in which are responsible for CAPS [44,45,46].
The study of the connections between autoimmunity and autoinflammation represents an exciting challenge for further studies [211,212,213,214]. Recently a study by Arakelyan and collegues, using a comparative gene expression analysis of a large set of transcriptome data, focused on finding existing similarities and common inflammatory components in ADs and autoinflammatory diseases [215]. The results revealed that some pathways were similarly perturbed in both diseases such as PI3K-Akt, Toll-like receptor, and NF-kappa β signalling, all important signals involved in immune cell polarisation, migration, growth, survival and differentiation [215]. They identified two clusters of diseases basing on specific disregulated pathways: one prevalently composed by autoimmune and the other by autoinflammatory disease [215]. Interestingly, some diseases did not seem to belong to the cluster they classically were believed to belong to. PAPA, one of the autoinflammatory diseases discussed above, was closer to the cluster of autoimmune diseases [215], conferming some differences found between this disease and the others autoinflammatory syndromes, such as the poor response to IL-1 blockade [209, 215]. Furthermore, SLE, a typical autoimmune disease, seemed to belong to the cluster of autoinflammatory syndromes [215], thus apparently remarking the systemic inflammatory nature of SLE and, as discussed above, a possible role for inflammasomes in its pathogenesis. This study identified disease specific variations in activation of common pathways, thus highliting not only the similarities but also the basic differences between the two types of diseases. The inflammatory aspects and the role of innate immunity in ADs should be further studied in order to identify their underlying autoinflammatory pathogenesis, thus opening up new perspectives on the link between innate and adaptive immune mechanisms. This would be a useful step towards developing the best tailored treatment strategy.
References
Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 27:621–668
de Torre-Minguela C, Mesa Del Castillo P, Pelegrín P (2017) The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol 8:43
McGonagle D, McDermott MF (2006) A proposed classification of the immunological diseases. PLoS Med 3:e297
Ombrello MJ (2015) Advances in the genetically complex autoinflammatory diseases. Semin Immunopathol 37:403–406
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161
Zen M, Gatto M, Domeneghetti M, Palma L, Borella E, Iaccarino L, Punzi L, Doria A (2013) Clinical guidelines and definitions of autoinflammatory diseases: contrasts and comparisons with autoimmunity-a comprehensive review. Clin Rev Allergy Immunol 45:227–235
Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, Ghirardello A, Iaccarino L, Punzi L (2012) Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev 12:22–30
Borella E, Palma L, Zen M, Bettio S, Nalotto L, Gatto M, Domeneghetti M, Iaccarino L, Punzi L, Doria A (2014) The body against self: autoinflammation and autoimmunity. Isr Med Assoc J 16:608–610
Davidson A, Diamond B (2001) Autoimmune diseases. N Engl J Med 345:340–350
Shi G, Zhang J, Zhang ZJ, Zhang X (2015) Systemic autoimmune diseases 2014. J Immunol Res 2015:183591
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426
Theofilopoulos AN, Gonzalez-Quintial R, Lawson BR, Koh YT, Stern ME, Kono DH, Beutler B, Baccala R (2010) Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol 6:146–156
Vajjhala PR, Ve T, Bentham A, Stacey KJ, Kobe B (2017) The molecular mechanisms of signaling by cooperative assembly formation in innate immunity pathways. Mol Immunol 86:23–37
McGonagle D, Savic S, McDermott MF (2007) The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Semin Immunopathol 29:303–313
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A (2002) Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature 416:603–607
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A (2005) RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J Exp Med 202:1171–1177
Kawasaki T, Kawai T, Akira S (2011) Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev 243:61–73
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A (2003) Activation of autoreactive B cells by CpG dsDNA. Immunity 19:837–847
Xie H, Kong X, Zhou H, Xie Y, Sheng L, Wang T, Xia L, Yan J (2015) TLR4 is involved in the pathogenic effects observed in a murine model of antiphospholipid syndrome. Clin Immunol 160:198–210
Cheng S, Wang H, Zhou H (2016) The role of TLR4 on B cell activation and anti-β(2)GPI antibody production in the antiphospholipid syndrome. J Immunol Res 2016:1719720
Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL (1999) Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med 190:1697–1710
Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J (1999) BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 189:1747–1756
Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F (2000) BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med 192:1453–1466
Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30:576–587
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Génin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, André F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178
Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T, Germain RN (2012) A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150:1235–1248
Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner-Hurevich M, Berzofsky JA, Nir-Paz R, Paul WE (2013) IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med 210:491–502
Lowenthal JW, Cerottini JC, MacDonald HR (1986) Interleukin 1-dependent induction of both interleukin 2 secretion and interleukin 2 receptor expression by thymoma cells. J Immunol 137:1226–1231
Koyama N, Harada N, Takahashi T, Mita S, Okamura H, Tominaga A, Takatsu K (1988) Role of recombinant interleukin-1 compared to recombinant T-cell replacing factor/interleukin-5 in B-cell differentiation. Immunology 63:277–283
Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, Lazova R, Kang I (2013) Self double-stranded (ds)DNA induces IL-1β production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J Immunol 190:1407–1415
Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN (2006) The comparative efficacy and safety of biologics for the treatment of rheumatoid arthritis: a systematic review and metaanalysis. J Rheumatol 33:2398–2340
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307:731–734
Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L (2012) Blau syndrome, clinical and genetic aspects. Autoimmun Rev 12:44–51
Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, Sfriso P, Verrecchia E, Tognon S, Bascherini V, Galeazzi M, Punzi L, Cantarini L (2014) Caveats and truths in genetic, clinical, autoimmune and autoinflammatory issues in Blau syndrome and early onset sarcoidosis. Autoimmun Rev 13:1220–1229
Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE (2007) The nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol 29:289–301
Caso F, Wouters CH, Rose CD, Costa L, Tognon S, Sfriso P, Cantarini L, Rigante D, Punzi L (2014) Blau syndrome and latent tubercular infection: an unresolved partnership. Int J Rheum Dis 17:586–587
Shinkai K, McCalmont TH, Leslie KS (2008) Cryopyrin-associated periodic syndromes and autoinflammation. Clin Exp Dermatol 33:1–9
Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, Kang I (2012) U1-small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol 188:4769–4775
Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ (2011) Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol 187:6143–6156
Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J, Cook HT, Kemper C (2013) C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122:3473–3481
Hui Z, Rong F, Chaohuan G, Yuefang H, Hongyue W, Shuang W, Jijun Z, Niansheng Y (2016) Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med 14:156
Rong F, Chaohuan G, Shuang W, Yuefang H, Ou J, Haoqiang H, Jingxian C, Bihua X, Mianjing Z, Jijun Z, Sun-sang J, Hongyang W, Felicia G, Niansheng Y, Shu Man F (2017) Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol 69:1636–1646
Caso F, Cantarini L, Lucherini OM, Sfriso P, Fioretti M, Costa L, Vitale A, Atteno M, Galeazzi M, Muscari I, Magnotti F, Frediani B, Punzi L, Rigante D (2014) Working the endless puzzle of hereditary autoinflammatory disorders. Mod Rheumatol 24:381–389
Caso F, Rigante D, Vitale A, Lucherini OM, Costa L, Atteno M, Compagnone A, Caso P, Frediani B, Galeazzi M, Punzi L, Cantarini L (2013) Monogenic autoinflammatory syndromes: state of the art on genetic, clinical, and therapeutic issues. Int J Rheumatol 2013:513782
Llndor NM, Arsenault TM, Solomon H, Seidman CE, McEvov MT (1997) A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc 72:611–615
Hutton HL, Ooi JD, Holdsworth SR, Kitching AR (2016) The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 21:736–744
Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, al-Gharbawy F, Majeed SA, al-Gazzar AH (1989) Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J Pediatr 115:730–734
Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, Ferguson PJ (2013) Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis 72:410–413
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgård U, Cowen EW, Pham TH, Booty M, Estes JD, Sandler NG, Plass N, Stone DL, Turner ML, Hill S, Butman JA, Schneider R, Babyn P, El-Shanti HI, Pope E, Barron K, Bing X, Laurence A, Lee CC, Chapelle D, Clarke GI, Ohson K, Nicholson M, Gadina M, Yang B, Korman BD, Gregersen PK, van Hagen PM, Hak AE, Huizing M, Rahman P, Douek DC, Remmers EF, Kastner DL, Goldbach-Mansky R (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 360:2426–2437
Pathak S, McDermott MF, Savic S (2017) Autoinflammatory diseases: update on classification diagnosis and management. J Clin Pathol 70:1–8
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee CC, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I (2014) Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 370:911–920
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M, Voth I, Hashkes PJ, Harel L, Hoffer V, Ling E, Yalcinkaya F, Kasapcopur O, Lee MK, Klevit RE, Renbaum P, Weinberg-Shukron A, Sener EF, Schormair B, Zeligson S, Marek-Yagel D, Strom TM, Shohat M, Singer A, Rubinow A, Pras E, Winkelmann J, Tekin M, Anikster Y, King MC, Levy-Lahad E (2014) Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 370:921–931
Lee-Kirsch MA, Wolf C, Kretschmer S, Roers A (2015) Type I interferonopathies—an expanding disease spectrum of immunodysregulation. Semin Immunopathol 37:349–357
Volpi S, Picco P, Caorsi R, Candotti F, Gattorno M (2016) Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J 14:35
Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D'Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ (2007) Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet 81:713–725
Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martínez de Villarreal L, dos Santos HG, Garg A (2010) PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet 87:866–872
Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, Toyoshima Y, Takahashi H, Standley DM, Tanaka K, Hamazaki J, Murata S, Obara K, Toyoshima I, Yasutomo K (2011) A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest 121:4150–4160
Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, Zhang X, Goldbach-Mansky R, Zlotogorski A (2012) Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 64:895–907
Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendínez F, Hernández A, López-Robledillo JC, Dadban A, Requena L, Paller AS (2010) Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol 62:489–495
Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, West B, Podoltsev NA, Boggon TJ, Kazmierczak BI, Lifton RP (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46:1135–1139
Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O'Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM, Goldbach-Mansky R (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46:1140–1146
Zhao Y, Shao F (2015) The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol Rev 265:85–102
Kofoed EM, Vance RE (2012) NAIPs: building an innate immune barrier against bacterial pathogens. NAIPs function as sensors that initiate innate immunity by detection of bacterial proteins in the host cell cytosol. Bioessays 34:589–598
Cush JJ (2013) Autoinflammatory syndromes. Dermatol Clin 31:471–480
Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD (2008) Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet 4:e1000024
Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC Jr, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S (2004) Gene map of the extended human MHC. Nat Rev Genet 5:889–899
Sollid LM, Pos W, Wucherpfennig KW (2014) Molecular mechanisms for contribution of MHC molecules to autoimmune diseases. Curr Opin Immunol 31:24–30
Morris DL, Taylor KE, Fernando MM, Nititham J, Alarcón-Riquelme ME, Barcellos LF, Behrens TW, Cotsapas C, Gaffney PM, Graham RR, Pons-Estel BA, Gregersen PK, Harley JB, Hauser SL, Hom G, International MHC and Autoimmunity Genetics Network, Langefeld CD, Noble JA, Rioux JD, Seldin MF, Systemic Lupus Erythematosus Genetics Consortium, Criswell LA, Vyse TJ (2012) Unraveling multiple MHC gene associations with systemic lupus erythematosus: model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am J Hum Genet 91:778–793
Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, Hulme J, Maier LM, Smyth D, Bailey R, Cooper JD, Ribas G, Campbell RD, Clayton DG, Todd JA, Wellcome Trust Case Control Consortium (2007) Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 450:887–892
Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA, Raj T, IMSGC.; ANZgene, Gourraud PA, Stranger BE, Oksenberg J, Olsson T, Taylor BV, Sawcer S, Hafler DA, Carrington M, De Jager PL, de Bakker PI (2013) Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet e1003926:9
Björck S, Brundin C, Lörinc E, Lynch KF, Agardh D (2010) Screening detects a high proportion of celiac disease in young HLA-genotyped children. J Pediatr Gastroenterol Nutr 50:49–53
Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest 101:273–281
Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, Simon M, Senshu T, Masson-Bessière C, Jolivet-Reynaud C, Jolivet M, Serre G (1999) The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol 162:585–594
Moeez S, John P, Bhatti A (2013) Anti-citrullinated protein antibodies: role in pathogenesis of RA and potential as a diagnostic tool. Rheumatol Int 33:1669–1673
Klareskog L, Padyukov L, Rönnelid J, Alfredsson L (2006) Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol 18:650–655
van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, Gomez-Touriño I, Arif S, Peakman M, Drijfhout JW, Roep BO (2014) Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes 63:237–247
Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, Ozyazgan Y, Sacli FS, Erer B, Inoko H, Emrence Z, Cakar A, Abaci N, Ustek D, Satorius C, Ueda A, Takeno M, Kim Y, Wood GM, Ombrello MJ, Meguro A, Gül A, Remmers EF, Kastner DL (2013) Genome-wide association analysis identifies new susceptibility loci for Behçet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet 45:202–207
Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, Oppermann U, Dilthey A, Pirinen M, Stone MA, Appleton L, Moutsianas L, Leslie S, Wordsworth T, Kenna TJ, Karaderi T, Thomas GP, Ward MM, Weisman MH, Farrar C, Bradbury LA, Danoy P, Inman RD, Maksymowych W, Gladman D, Rahman P, Spondyloarthritis Research Consortium of Canada (SPARCC), Morgan A, Marzo-Ortega H, Bowness P, Gaffney K, Gaston JS, Smith M, Bruges-Armas J, Couto AR, Sorrentino R, Paladini F, Ferreira MA, Xu H, Liu Y, Jiang L, Lopez-Larrea C, Díaz-Peña R, López-Vázquez A, Zayats T, Band G, Bellenguez C, Blackburn H, Blackwell JM, Bramon E, Bumpstead SJ, Casas JP, Corvin A, Craddock N, Deloukas P, Dronov S, Duncanson A, Edkins S, Freeman C, Gillman M, Gray E, Gwilliam R, Hammond N, Hunt SE, Jankowski J, Jayakumar A, Langford C, Liddle J, Markus HS, Mathew CG, McCann OT, McCarthy MI, Palmer CN, Peltonen L, Plomin R, Potter SC, Rautanen A, Ravindrarajah R, Ricketts M, Samani N, Sawcer SJ, Strange A, Trembath RC, Viswanathan AC, Waller M, Weston P, Whittaker P, Widaa S, Wood NW, McVean G, Reveille JD, Wordsworth BP, Brown MA, Donnelly P, Australo-Anglo-American Spondyloarthritis Consortium (TASC); Wellcome Trust Case Control Consortium 2 (WTCCC2) (2011) Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet 43:761–767
Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, Siminovitch KA, Bae SC, Plenge RM, Gregersen PK, de Bakker PI (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44:291–296
Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T, Gudjonsson JE, Kang HM, Allen MH, McManus R, Novelli G, Samuelsson L, Schalkwijk J, Ståhle M, Burden AD, Smith CH, Cork MJ, Estivill X, Bowcock AM, Krueger GG, Weger W, Worthington J, Tazi-Ahnini R, Nestle FO, Hayday A, Hoffmann P, Winkelmann J, Wijmenga C, Langford C, Edkins S, Andrews R, Blackburn H, Strange A, Band G, Pearson RD, Vukcevic D, Spencer CC, Deloukas P, Mrowietz U, Schreiber S, Weidinger S, Koks S, Kingo K, Esko T, Metspalu A, Lim HW, Voorhees JJ, Weichenthal M, Wichmann HE, Chandran V, Rosen CF, Rahman P, Gladman DD, Griffiths CE, Reis A, Kere J, Collaborative Association Study of Psoriasis (CASP); Genetic Analysis of Psoriasis Consortium; Psoriasis Association Genetics Extension; Wellcome Trust Case Control Consortium 2, Nair RP, Franke A, Barker JN, Abecasis GR, Elder JT, Trembath RC (2012) Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 44:1341–1348
McGonagle D, Aziz A, Dickie LJ, McDermott MF (2009) An integrated classification of pediatric inflammatory diseases, based on the concepts of autoinflammation and the immunological disease continuum. Pediatr Res 65:38R–45R
Mohan VK, Ganesan N, Gopalakrishnan R (2014) Association of susceptible genetic markers and autoantibodies in rheumatoid arthritis. J Genet 93:597–605
Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, International Inflammatory Bowel Disease Genetics Consortium, Parkes M, Vermeire S, Rioux JD, Mansfield J, Silverberg MS, Radford-Smith G, McGovern DP, Barrett JC, Lees CW et al (2016) Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 387:156–167
Moroldo MB, Donnelly P, Saunders J, Glass DN, Giannini EH (1998) Transmission disequilibrium as a test of linkage and association between HLA alleles and pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum 41:1620–1624
Rossi-Semerano L, Koné-Paut I (2012) Is Still’s disease an autoinflammatory syndrome? Int J Inflamm 2012:480373
Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM (1996) Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96
Lebowitz MS, O'Herrin SM, Hamad AR, Fahmy T, Marguet D, Barnes NC, Pardoll D, Bieler JG, Schneck JP (1999) Soluble, high-affinity dimers of T-cell receptors and class II major histocompatibility complexes: biochemical probes for analysis and modulation of immune responses. Cell Immunol 192:175–184
Novak EJ, Liu AW, Nepom GT, Kwok WW (1999) MHC class II tetramers identify peptide-specific human CD4(+) T cells proliferating in response to influenza A antigen. J Clin Invest 104:R63–R67
Klein L, Kyewski B, Allen PM, Hogquist KA (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 14:377–391
Andrade LE (2009) Future perspective for diagnosis in autoimmune diseases. An Acad Bras Cienc 81:367–380
Worth A, Thrasher AJ, Gaspar HB (2006) Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol 133:124–140
Beretta L, Cappiello F, Barili M, Bertolotti F, Scorza R (2007) T-889C IL-1alpha promoter polymorphism influences the response to oral cyclophosphamide in scleroderma patients with alveolitis. Clin Rheumatol 26:88–91
Yang CA, Chiang BL (2015) Inflammasomes and human autoimmunity: a comprehensive review. J Autoimmun 61:1–8
dos Santos BP, Valverde JV, Rohr P, Monticielo OA, Brenol JC, Xavier RM, Chies JA (2012) TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 21:302–309
Zhang J, Zhu Q, Meng F, Lei H, Zhao Y (2014) Association study of TLR-9 polymorphisms and systemic lupus erythematosus in northern Chinese Han population. Gene 533:385–388
Huang CM, Huang PH, Chen CL, Lin YJ, Tsai CH, Huang WL, Tsai FJ (2012) Association of toll-like receptor 9 gene polymorphism in Chinese patients with systemic lupus erythematosus in Taiwan. Rheumatol Int 32:2105–2109
Piotrowski P, Lianeri M, Wudarski M, Olesińska M, Jagodziński PP (2013) Contribution of toll-like receptor 9 gene single-nucleotide polymorphism to systemic lupus erythematosus. Rheumatol Int 33:1121–1125
Oyanguren-Desez O, Rodríguez-Antigüedad A, Villoslada P, Domercq M, Alberdi E, Matute C (2011) Gain-of-function of P2X7 receptor gene variants in multiple sclerosis. Cell Calcium 50:468–472
Chen S, Sun B (2013) Negative regulation of NLRP3 inflammasome signaling. Protein Cell 4:251–258
Haneklaus M, O'Neill LA, Coll RC (2013) Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 25:40–45
Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA (2007) NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med 356:1216–1225
Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Söderkvist P (2008) Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology (Oxford) 47:415–417
Yang CA, Huang ST, Chiang BL (2014) Association of NLRP3 and CARD8 genetic polymorphisms with juvenile idiopathic arthritis in a Taiwanese population. Scand J Rheumatol 43:146–152
Yang CA, Huang ST, Chiang BL (2015) Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology (Oxford) 54:324–331
Maggadottir SM, Sullivan KE (2014) The intersection of immune deficiency and autoimmunity. Curr Opin Rheumatol 26:570–578
Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Fischer A, Franco JL, Geha RS, Hammarström L, Nonoyama S, Notarangelo LD, Ochs HD, Puck JM, Roifman CM, Seger R, Tang ML (2011) Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2:54
Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC (2003) Aire regulates negative selection of organ-specific T cells. Nat Immunol 4:350–354
Kisand K, Peterson P, Laan M (2014) Lymphopenia-induced proliferation in aire-deficient mice helps to explain their autoimmunity and differences from human patients. Front Immunol 5:51
Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG (2006) Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 116:1713–1722
Bonhomme D, Hammarström L, Webster D, Chapel H, Hermine O, Le Deist F, Lepage E, Romeo PH, Levy Y (2000) Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol 165:4725–4730
Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, Eibel H, Schlesier M, Peter HH (2002) Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 99:1544–1551
Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C (2008) Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol 128:314–321
Notarangelo LD, Miao CH, Ochs HD (2008) Wiskott-Aldrich syndrome. Curr Opin Hematol 15:30–36
Castiello MC, Bosticardo M, Pala F, Catucci M, Chamberlain N, van Zelm MC, Driessen GJ, Pac M, Bernatowska E, Scaramuzza S, Aiuti A, Sauer AV, Traggiai E, Meffre E, Villa A, van der Burg M (2014) Wiskott-Aldrich syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J Autoimmun 50:42–50
Westerberg LS, Klein C, Snapper SB (2008) Breakdown of T cell tolerance and autoimmunity in primary immunodeficiency—lessons learned from monogenic disorders in mice and men. Curr Opin Immunol 20:646–654
Cravens PD, Lipsky PE (2002) Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol Cell Biol 80:497–505
Steinman RM (1991) The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271–296
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K (2000) Immunobiology of dendritic cells. Annu Rev Immunol 18:767–811
Ludewig B, Junt T, Hengartner H, Zinkernagel RM (2001) Dendritic cells in autoimmune diseases. Curr Opin Immunol 13:657–662
Corti D, Sallusto F, Lanzavecchia A (2011) High throughput cellular screens to interrogate the human T and B cell repertoires. Curr Opin Immunol 23:430–435
Leyendeckers H, Odendahl M, Löhndorf A, Irsch J, Spangfort M, Miltenyi S, Hunzelmann N, Assenmacher M, Radbruch A, Schmitz J (1999) Correlation analysis between frequencies of circulating antigen-specific IgG-bearing memory B cells and serum titers of antigen-specific IgG. Eur J Immunol 29:1406–1417
Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, Busch DH (2007) A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity 27:985–997
Amanna IJ, Carlson NE, Slifka MK (2007) Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med 357:1903–1915
Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A (2012) Integration of B cell responses through toll-like receptors and antigen receptors. Nat Rev Immunol 12:282–294
Leadbetter EA, Rifkin IR, Marshak-Rothstein A (2003) Toll-like receptors and activation of autoreactive B cells. Curr Dir Autoimmun 6:105–122
Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF (2007) TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol 178:2415–2420
Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF (2009) The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol 182:7459–7472
Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, DeFranco AL (2011) Selective utilization of toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity 34:375–384
Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125:2194–2202
Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV (2006) TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203:553–561
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ (2006) Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25:417–428
Hua Z, Gross AJ, Lamagna C, Ramos-Hernández N, Scapini P, Ji M, Shao H, Lowell CA, Hou B, DeFranco AL (2014) Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 192:875–885
Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F (2007) BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med 204:1959–1971
Suurmond J, Rivellese F, Dorjée AL, Bakker AM, Rombouts YJ, Rispens T, Wolbink G, Zaldumbide A, Hoeben RC, Huizinga TW, Toes RE (2015) Toll-like receptor triggering augments activation of human mast cells by anti-citrullinated protein antibodies. Ann Rheum Dis 74:1915–1923
Pitzalis C, Jones GW, Bombardieri M, Jones SA (2014) Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 14:447–462
Thapa DR, Tonikian R, Sun C, Liu M, Dearth A, Petri M, Pepin F, Emerson RO, Ranger A (2015) Longitudinal analysis of peripheral blood T cell receptor diversity in patients with systemic lupus erythematosus by next-generation sequencing. Arthritis Res Ther 17:132
Attaf M, Huseby E, Sewell AK (2015) αβ T cell receptors as predictors of health and disease. Cell Mol Immunol 12:391–399
Raulet DH (1989) The structure, function, and molecular genetics of the gamma/delta T cell receptor. Annu Rev Immunol 7:175–207
Chothia C, Boswell DR, Lesk AM (1988) The outline structure of the T-cell alpha beta receptor. EMBO J 7:3745–3755
Alam SM, Davies GM, Lin CM, Zal T, Nasholds W, Jameson SC, Hogquist KA, Gascoigne NR, Travers PJ (1999) Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity 10:227–237
Almeida A, Rocha B, Freitas AA, Tanchot C (2005) Homeostasis of T cell numbers: from thymus production to peripheral compartmentalization and the indexation of regulatory T cells. Semin Immunol 17:239–249
Takeuchi T, Abe T, Koide J, Hosono O, Morimoto C, Homma M (1984) Cellular mechanism of DNA-specific antibody synthesis by lymphocytes from systemic lupus erythematosus patients. Arthritis Rheum 27:766–773
Datta SK, Kaliyaperumal A, Mohan C, Desai-Mehta A (1997) T helper cells driving pathogenic anti-DNA autoantibody production in lupus: nucleosomal epitopes and CD40 ligand signals. Lupus 6:333–336
Shivakumar S, Tsokos GC, Datta SK (1989) T cell receptor alpha/beta expressing double-negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol 143:103–112
Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M (1990) Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med 171:265–292
Desai-Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK (1995) Structure and specificity of T cell receptors expressed by potentially pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest 95:531–541
Luo W, Ma L, Wen Q, Wang N, Zhou MQ, Wang XN (2008) Analysis of the interindividual conservation of T cell receptor alpha- and beta-chain variable regions gene in the peripheral blood of patients with systemic lupus erythematosus. Clin Exp Immunol 154:316–324
Winchester R, Wiesendanger M, Zhang HZ, Steshenko V, Peterson K, Geraldino-Pardilla L, Ruiz-Vazquez E, D'Agati V (2012) Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+ T cell β-chain clonotypes in progressive lupus nephritis. Arthritis Rheum 64:1589–1600
Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L (2015) Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 7:274ra18
Pearce EL, Poffenberger MC, Chang CH, Jones RG (2013) Fueling immunity: insights into metabolism and lymphocyte function. Science 342:1242454
Diveu C, McGeachy MJ, Cua DJ (2008) Cytokines that regulate autoimmunity. Curr Opin Immunol 20:663–668
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S (2007) Phenotypic and functional features of human Th17 cells. J Exp Med 204:1849–1861
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8:950–957
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F (2012) Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484:514–518
Noster R, de Koning HD, Maier E, Prelog M, Lainka E, Zielinski CE (2016) Dysregulation of proinflammatory versus anti-inflammatory human T(H)17 cell functionalities in the autoinflammatory Schnitzler syndrome. J Allergy Clin Immunol 138:1161–1169.e6
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24:179–189
Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG (1996) Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol 156:5–7
Chevalier N, Thorburn AN, Macia L, Tan J, Juglair L, Yagita H, Yu D, Hansbro PM, Mackay CR (2014) Inflammation and lymphopenia trigger autoimmunity by suppression of IL-2-controlled regulatory T cell and increase of IL-21-mediated effector T cell expansion. J Immunol 193:4845–4858
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20:62–68
Kuhn A, Beissert S, Krammer PH (2009) CD4(+)CD25 (+) regulatory T cells in human lupus erythematosus. Arch Dermatol Res 301:71–81
Sakaguchi S, Sakaguchi N (2005) Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol 24:211–226
Sakaguchi S (2004) Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 22:531–562
Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T (2001) Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev 182:18–32
Shevach EM (2002) CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol 2:389–400
Barath S, Aleksza M, Tarr T, Sipka S, Szegedi G, Kiss E (2007) Measurement of natural (CD4+CD25high) and inducible (CD4+IL-10+) regulatory T cells in patients with systemic lupus erythematosus. Lupus 16:489–496
Cao D, Malmström V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C (2003) Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol 33:215–223
Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, Perricone R (2013) Complement and autoimmunity. Immunol Res 56:477–491
Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM (2005) Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett 97:171–179
Nauta AJ, Roos A, Daha MR (2004) A regulatory role for complement in innate immunity and autoimmunity. Int Arch Allergy Immunol 134:310–323
Morgan BP, Harris CL (2003) Complement therapeutics; history and current progress. Mol Immunol 40:159–170
Avery TY, van de Cruys M, Austen J, Stals F, Damoiseaux JG (2014) Anti-nuclear antibodies in daily clinical practice: prevalence in primary, secondary, and tertiary care. J Immunol Res 2014:401739
Kumar Y, Bhatia A, Minz RW (2009) Antinuclear antibodies and their detection methods in diagnosis of connective tissue diseases: a journey revisited. Diagn Pathol 4:1
Kovarik J (2013) From immunosuppression to immunomodulation: current principles and future strategies. Pathobiology 80:275–281
Baker KF, Isaacs JD (2014) Prospects for therapeutic tolerance in humans. Curr Opin Rheumatol 26:219–227
Löwenberg M, Stahn C, Hommes DW, Buttgereit F (2008) Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids 73:1025–1029
Caso F, Lubrano E, Del Puente A, Caso P, Peluso R, Foglia F, Benigno C, Girolimetto N, Bottiglieri P, Scarpa R, Costa L (2016) Progress in understanding and utilizing TNF-α inhibition for the treatment of psoriatic arthritis. Expert Rev Clin Immunol 12:315–331
Caso F, Del Puente A, Peluso R, Caso P, Girolimetto N, Del Puente A, Scarpa R, Costa L (2016) Emerging drugs for psoriatic arthritis. Expert Opin Emerg Drugs 21:69–79
Akgul O, Kilic E, Kilic G, Ozgocmen S (2013) Efficacy and safety of biologic treatments in familial Mediterranean fever. Am J Med Sci 346:137–141
Vitale A, Rigante D, Lucherini OM, Caso F, Muscari I, Magnotti F, Brizi MG, Guerrini S, Patti M, Punzi L, Galeazzi M, Cantarini L (2013) Biological treatments: new weapons in the management of monogenic autoinflammatory disorders. Mediat Inflamm 2013:939847
Cantarini L, Rigante D, Merlini G, Vitale A, Caso F, Lucherini OM, Sfriso P, Frediani B, Punzi L, Galeazzi M, Cimaz R, Obici L (2014) The expanding spectrum of low-penetrance TNFRSF1A gene variants in adults presenting with recurrent inflammatory attacks: clinical manifestations and long-term follow-up. Semin Arthritis Rheum 43:818–823
Pascual-Salcedo D, Plasencia C, Ramiro S, Nuño L, Bonilla G, Nagore D, Ruiz Del Agua A, Martínez A, Aarden L, Martín-Mola E, Balsa A (2011) Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology (Oxford) 50:1445–1452
Afif W, Loftus EV Jr, Faubion WA, Kane SV, Bruining DH, Hanson KA, Sandborn WJ (2010) Clinical utility of measuring infliximab and human anti-chimeric antibody concentrations in patients with inflammatory bowel disease. Am J Gastroenterol 105:1133–1139
Dinarello CA, van der Meer JW (2013) Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 25:469–484
Sfriso P, Priori R, Valesini G, Rossi S, Montecucco CM, D'Ascanio A, Carli L, Bombardieri S, LaSelva G, Iannone F, Lapadula G, Alivernini S, Ferraccioli G, Colaci M, Ferri C, Iacono D, Valentini G, Costa L, Scarpa R, LoMonaco A, Bagnari V, Govoni M, Piazza I, Adami S, Ciccia F, Triolo G, Alessandri E, Cutolo M, Cantarini L, Galeazzi M, Ruscitti P, Giacomelli R, Caso F, Galozzi P, Punzi L (2016) Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol 35:1683–1689
Vaitla PM, Radford PM, Tighe PJ, Powell RJ, McDermott EM, Todd I, Drewe E (2011) Role of interleukin-6 in a patient with tumor necrosis factor receptor-associated periodic syndrome: assessment of outcomes following treatment with the anti-interleukin-6 receptor monoclonal antibody tocilizumab. Arthritis Rheum 63:1151–1155
Fonseca JE, Santos MJ, Canhão H, Choy E (2009) Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev 8:538–542
Henrickson M, Wang H (2017) Tocilizumab reverses cerebral vasculopathy in a patient with homozygous SAMHD1 mutation. Clin Rheumatol 36:1445–1451
Woodrick RS, Ruderman EM (2012) IL-6 inhibition for the treatment of rheumatoid arthritis and other conditions. Bull NYU Hosp Jt Dis 70:195–199
Caso F, Costa L, Rigante D, Lucherini OM, Caso P, Bascherini V, Frediani B, Cimaz R, Marrani E, Nieves-Martín L, Atteno M, Raffaele CG, Tarantino G, Galeazzi M, Punzi L, Cantarini L (2014) Biological treatments in Behçet’s disease: beyond anti-TNF therapy. Mediat Inflamm 2014:107421
Croft M, Siegel RM (2017) Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol 13:217–233
Greco E, Aita A, Galozzi P, Gava A, Sfriso P, Negm OH, Tighe P, Caso F, Navaglia F, Dazzo E, De Bortoli M, Rampazzo A, Obici L, Donadei S, Merlini G, Plebani M, Todd I, Basso D, Punzi L (2015) The novel S59P mutation in the TNFRSF1A gene identified in an adult onset TNF receptor associated periodic syndrome (TRAPS) constitutively activates NF-κB pathway. Arthritis Res Ther 17:93
Manzi S, Sánchez-Guerrero J, Merrill JT, Furie R, Gladman D, Navarra SV, Ginzler EM, D’Cruz DP, Doria A, Cooper S, Zhong ZJ, Hough D, Freimuth W, Petri MA, BLISS-52 and BLISS-76 Study Groups (2012) Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis 71:1833–1838
Leget GA, Czuczman MS (1998) Use of rituximab, the new FDA-approved antibody. Curr Opin Oncol 10:548–551
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T (2004) Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 350:2572–2581
Andrade-Ortega L, Irazoque-Palazuelos F, Muñóz-López S, Rosales-Don Pablo VM (2013) Efficacy and tolerability of rituximab in patients with rhupus. Reumatol Clin 9:201–205
Caso F, Fiocco U, Costa L, Sfriso P, Punzi L, Doria A (2014) Successful use of rituximab in a young patient with immunoglobulin G4-related disease and refractory scleritis. Joint Bone Spine 81:190–192
Shin JI, Eisenhut M (2014) A beneficial effect of rituximab on autoimmune thrombotic thrombocytopenic purpura: just a B-cell depletion? J Allergy Clin Immunol 133:600
McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, Dahmen G, Wollenhaupt J, Schulze-Koops H, Kogan J, Ma S, Schumacher MM, Bertolino AP, Hueber W, Tak PP (2014) Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 73:349–356
Kavanaugh A, Ritchlin C, Rahman P, Puig L, Gottlieb AB, Li S, Wang Y, Noonan L, Brodmerkel C, Song M, Mendelsohn AM, McInnes IB, PSUMMIT-1 and 2 Study Groups (2014) Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis 73:1000–1006
Östör AJK (2008) Abatacept: a T-cell co- stimulation modulator for the treatment of rheumatoid arthritis. Clin Rheumatol 28:1343–1353
Scarpa R, Costa L, Atteno M, Del Puente A, Caso F, Moll JM (2013) Psoriatic arthritis: advances in pharmacotherapy based on molecular target. Expert Opin Pharmacother 14:2311–2313
Strand V, Kavanaugh AF (2004) The role of interleukin-1 in bone resorption in rheumatoid arthritis. Rheumatology (Oxford) 43:10–16
Demidowich AP, Freeman AF, Kuhns DB, Aksentijevich I, Gallin JI, Turner ML, Kastner DL, Holland SM (2012) Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum 64:2022–2027
Nepom GT (2012) MHC class II tetramers. J Immunol 188:2477–2482
Kappler J, Kubo R, Haskins K, Hannum C, Marrack P, Pigeon M, McIntyre B, Allison J, Trowbridge I (1983) The major histocompatibility complex-restricted antigen receptor on T cells in mouse and man: identification of constant and variable peptides. Cell 35:295–302
Damoiseaux JG, Tervaert JW (2006) From ANA to ENA: how to proceed? Autoimmun Rev 5:10–17
Wiik AS, Gordon TP, Kavanaugh AF, Lahita RG, Reeves W, van Venrooij WJ, Wilson MR, Fritzler M, IUIS/WHO/AF/CDC Committee for the Standardization of Autoantibodies in Rheumatic and Related Diseases (2004) Cutting edge diagnostics in rheumatology: the role of patients, clinicians, and laboratory scientists in optimizing the use of autoimmune serology. Arthritis Rheum 51:291–298
Caso F, Iaccarino L, Bettio S, Ometto F, Costa L, Punzi L, Doria A (2013) Refractory pemphigus foliaceus and Behçet’s disease successfully treated with tocilizumab. Immunol Res 56:390–397
Arakelyan A, Nersisyan L, Poghosyan D, Khondkaryan L, Hakobyan A, Löffler-Wirth H, Melanitou E, Binder H (2017) Autoimmunity and autoinflammation: a systems view on signaling pathway dysregulation profiles. PLoS One 12:e0187572
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Rights and permissions
About this article

Cite this article
Caso, F., Costa, L., Nucera, V. et al. From autoinflammation to autoimmunity: old and recent findings. Clin Rheumatol 37, 2305–2321 (2018). https://doi.org/10.1007/s10067-018-4209-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4209-9