
Abstract
Adult-onset Still’s disease (AOSD) is a systemic inflammatory condition of unknown aetiology characterized by typical episodes of spiking fever, evanescent rash, arthralgia, leukocytosis and hyperferritinemia. Given the lack of data in Italian series, we promote a multicentric data collection to characterize the clinical phenotype of Italian patients with AOSD. Data from 245 subjects diagnosed with AOSD were collected by 15 centres between March and May 2013. The diagnosis was made following Yamaguchi’s criteria. Data regarding clinical manifestations, laboratory features, disease course and treatments were reported and compared with those presented in other published series of different ethnicity. The most frequent features were the following: arthritis (93 %), pyrexia (92.6 %), leukocytosis (89 %), negative ANA (90.4 %) and neutrophilia (82 %). As compared to other North American, North European, Middle Eastern and Far Eastern cohorts, Italian data show differences in clinical and laboratory findings. Regarding the treatments, in 21.9 % of cases, corticosteroids and traditional DMARDs have not been able to control the disease while biologics have been shown to be effective in 48 to 58 patients. This retrospective work summarizes the largest Italian multicentre series of AOSD patients and presents clinical and laboratory features that appear to be influenced by the ethnicity of the affected subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult-onset Still’s disease (AOSD) is a rare complex autoinflammatory disorder of unknown aetiology. First described in children by George Still in 1896 [1], same symptoms were reported in 1971 by Eric Bywaters in adult patients who did not fulfil criteria for classic rheumatoid arthritis [2]. Its incidence is approximately 0.16 per 100,000 persons in France [3], and it affects usually young adults (median age 36 years).
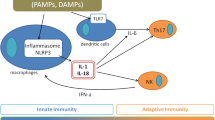
The pathogenesis of AOSD is unknown, but genetic factors and various infectious agents have been considered as predisposing factors [4]. Genetically predisposed individuals would develop autoinflammatory reactions to environmental triggers, leading to neutrophil and macrophage activation, a hallmark of AOSD.
Main features of AOSD include high spiking fever, arthralgia or arthritis, evanescent skin rash, sore throat, hepatosplenomegaly, leukocytosis with neutrophilia, elevated liver enzymes and ESR, and hyperferritinemia with decreased glycosylated ferritin (<20 %). According to the clinical course of the disease, AOSD may be conventionally divided into three main patterns [5]: monocyclic pattern, characterized by systemic single episode completely resolving within months; intermittent or polycyclic pattern, associated with one or more disease flares and characterized by complete remissions lasting to a couple of years; and chronic pattern, usually associated with polyarthritis.
Diagnosis is difficult, delayed and usually based on classification criteria set, such as Yamaguchi and Fautrel’s criteria [5, 6]. Due to the heightened clinical heterogeneity in AOSD, exclusion of other entities including infectious, neoplastic and autoimmune disorders should be ruled out before any diagnosis.
The treatment of AOSD remains largely empirical, relying only on small retrospective case series [7]. Patients usually respond to nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, disease-modifying anti-rheumatic drugs (DMARDs) such as methotrexate, and polyvalent intravenous immunoglobulins (IVIg). Biologic agents are considered for treatment of corticosteroids- and DMARDs-refractory cases and represent major therapeutic advances. IL-1 inhibition with Anakinra (IL-1 receptor antagonist), Canakinumab (anti-IL-1 monoclonal antibody) or Rilonacept (anti-IL-1 fusion protein) seem to be effective, well tolerated and steroid-sparing in systemic AOSD patients, whereas TNF-α blockers could be interesting in chronic polyarticular AOSD. Inhibition of IL-6 with tocilizumab is documented and seems effective in patients with active arthritis [8].
Most of the data on AOSD come from monocentric studies with limited number of cases.
The Italian Society of Rheumatology study group on autoinflammatory diseases promoted a multicentric data collection of Italian AOSD cases aiming to develop a national registry. In this work, we report the clinical manifestations, laboratory profile, patterns of disease course and therapy, collected by the different centres.
Patients and methods
Fifteen Italian University Hospital centres participated in this retrospective study and collected clinical and laboratory data together with disease and therapy information from AOSD patients. Data is referred to a cohort of patients recorded between March and May 2013. Diagnosis of AOSD was based on Yamaguchi’s diagnostic criteria [6] and required the presence of 5 major, minor or exclusion findings, of whom 2 or more must be major. Major Yamaguchi criteria include the presence of intermittent high spiking fever (≥39 °C) lasting 1 week or longer, arthralgia lasting more than 2 weeks, characteristic rash and leukocytosis (white blood cell count ≥10,000/μL per mm3) with >80 % granulocytes, whereas minor criteria are sore throat, lymphadenopathy and/or splenomegaly, abnormal liver function tests and negative rheumatoid factor and anti-nuclear autoantibody (ANA) titre. The presence of infections, malignancies and other rheumatic diseases are exclusion conditions.
Each centre has recorded patients’ data by using a standardized form, created specifically for collecting AOSD cases. Together with clinical characteristics, laboratory features are reported including blood cell count, coagulation parameters, serum ferritin (SF), ESR, C-reactive protein (CRP), liver enzymes, rheumatoid factor (RF) and ANA.
All treatments were listed with information about doses, durations and adverse events, along with the follow-up of different biologic treatments.
Concerning the disease course, the patients were stratified into four classes: polycyclic systemic, chronic articular polycyclic, monocyclic systemic and chronic articular monocyclic.
The collected data were compared with those reported in five published series of different ethnic origin and with sample size more than 50 subjects [4, 5, 8–10]. Descriptive statistics are represented as mean ± standard deviation (SD).
This retrospective study was approved by Padua Hospital Ethics Committee.
Results
Clinical features
Two hundred and forty-five patients (116 females and 129 males) with AOSD diagnosis and Caucasian origin were collected. The median age at the onset of disease manifestations was 38.8 years (range, 16–78.6), and the median delay of diagnosis was 1.5 months (range, 0–232). The disease manifestations were reported and compared with other series [4, 5, 8–10] in Table 1. At the time of onset, principal manifestations consisted of arthralgia (93 %), arthritis (75.8 %), pyrexia (92.6 %) and leukocytosis (89 %). Patients experiencing fever report high temperature (39.1 ± 0.7 °C) and one or two spikes per day in 27.3 and 51.9 % of cases, respectively. The pattern of fever was intermittent, with an average duration of 10.3 days and an intercritical phase of 3.2 days. Other symptoms observed in AOSD patients were the following: typical rash (67.7 %), sore throat or pharyngitis (61.8 %) and lymphadenopathy (60.4 %). Among patients with arthritis, 23.7 % had monoarthritis, 44.3 % had oligoarthritis and 32.0 % had polyarthritis.
Reactive hemophagocytic syndrome (RHS) was reported in seven patients (2.85 %).
The clinical course in our patients were polycyclic systemic in 40.8 % of cases, chronic articularpolycyclic in 30.7 %, monocyclic systemic in 23.9 % and chronic articular monocyclic in 4.6 %.
Laboratory features
As presented in Table 2, leukocytosis (white blood cell count ≥10,000/mm3) occurs in 81 % of patients (mean ± SD, 16,200 ± 7850/mm3) and was composed of ≥80 % PMN in 70.3 % (mean ± SD, 13,483 ± 7341/mm3). CRP was increased in 93 % of patients (mean ± SD, 101.9 ± 88.6 mg/L) in a range 1–355 mg/L. Platelet count was 378 ± 176 × 109/L (range, 20–922), and thrombocytosis (>400 × 109/L) occurred in 46 % of patients.
Elevation of hepatic enzymes was observed in 53.5 % of patients, and rheumatoid factor was positive only in 3.8 %. Patients were tested for autoantibodies: 0.5 % of patients was positive for anti-CCP, 9.6 % for ANA and 3.7 % for anti-ENA. Serum ferritin above the normal levels was observed in 56.4 % of patients (mean ± SD, 5743 ± 9723) within a range of 156–52,395.
Comorbidities
At the time of the last follow-up, almost 70 % of patients presented comorbidities: cardiovascular diseases in 61 patients, pneumopathies in 17, nephritis in 12, hepatitis in 11, bowel diseases in 8, thyroiditis in 26, autoimmune diseases in 8, diabetes in 19, neoplasia in 6, and other diseases in 4 patients (Table 3).
Treatment
84.5 % of patients received steroids and 3 % steroids in bolus. Fifty-one percent of those patients took DMARDs and about 35 % concomitant drugs (FANS), such as indomethacin, ibuprofen and diclofenac. Among patients treated with DMARDs, 60 % used methotrexate, 18.5 % cyclosporin, 13.2 % hydroxychloroquine, 3.4 % salazopyrin, 2.4 % azathiopyrin and 2.4 % leflunomide.
As reported in Table 4, treatment with first-line biologic agents was prescribed on 58 occasions (23.7 %), 3.5 ± 4.3 years after the disease diagnosis and at 40.6 ± 15.3 years of age. Within them, 46.6 % received TNF-blockers and 53.4 % anakinra. Second-line biologic drugs were used 19 times (7.7 %), of which TNF-blockers in 10 patients, anakinra in 4 patients and others in 5 patients. Moreover, third-line biologic agents were used as treatment on 7 occasions (2.8 %): 4 times TNF-blockers and 3 times others.
Forty-two patients are reported to continue with biologic treatments; in details, 30 patients use first-line biologic agents (13 TNF-blockers and 17 anakinra), 9 patients take second-line agents (3 TNF-blockers, 4 anakinra and 2 others) and 3 patients receive third-line biologics (2 TNF-blockers and 1 others). Sixteen up to 58 patients treated with biologic agents have definitively withdrawn this treatment, due to adverse events (8 times), loss or lack of efficacy (2 times) and remission (6 times).
Given the absence of consensus regarding a disease activity measures in AOSD, the therapeutic efficacy of the biologic agents was evaluated based on disappearance of all clinical symptoms and biological manifestations (complete response, partial response, no response). The therapeutic efficacy of anakinra and TNF-blockers is reported in Table 5.
Discussion
AOSD is a heterogeneous complex disorder with unknown pathogenesis and difficult diagnosis [7]. Due to the limited number of clinical cases, often referring to individual centres, the Italian Society of Rheumatology study group on autoinflammatory diseases promoted a multicentric collection of Italian AOSD patients’ data. The main aims of this registry are to define the clinical and laboratory pattern of AOSD and the therapeutic attitude towards the disease in Italy. Furthermore, as long-term purpose, the registry aims to identify prognostic factors of the disease. Up to date, AOSD data on the Italian population derive only from monocentric studies and are not a complete representation of the nation [11–14].
In this retrospective study, we reported 245 patients with AOSD diagnosis referring to 15 Italian Rheumatologic Centres, 6 from northern Italy, 5 from central Italy and 4 from the southern Italy. To the best of our knowledge, this registry collects the largest number of AOSD patient data in Italy.
Tables 1 and 2 present a wide variability in the frequencies of clinical and laboratory features in our Italian AOSD patients compared with data from different ethnicity. Italian and Japanese patients were generally older at disease onset than subjects from the other series. In our study, the mean age of the patients at the time of diagnosis was 40.5 ± 16.5 years, whereas in other European countries, Turkey and China, the mean age is around 34 years. The delay to final diagnosis was highly variable from 0 to 232 months (median 1.5 months), which indicates possible diagnostic difficulties.
The tendency for AOSD to show female predominance has been already noticed [4, 5, 9, 10], but the reason is not clear. Curiously, in our population, males and females are equally affected; this may be due to an unexpected selection bias or may reflect a peculiar characteristic of the Italian population.
In the present study, arthralgia (93 %) and fever (92.6 %) were the most common clinical findings, the frequency of which was similar to the other reported cases. Symptoms such as pharyngitis and maculopapular rash appear to be less frequent in Italian and Turkish subjects, whereas lymphadenopathy is most experienced by Italian, Spanish and Chinese patients than other series. Spanish patients showed the lowest frequency of hepatomegaly and sore throat, while the French subjects had the highest frequency of pericarditis. Recently, in a single-centre retrospective study on 39 AOSD patients, F. Dall’Ara et al. looked for predictors of the use of biologic agents. They suggested that pericarditis may be a possible marker of severe AOSD associated with higher probability of a disease refractory to conventional DMARDs. They found pericarditis in 7 patients (38 %) receiving biologic agents and in 1 patient (5 %) of those receiving traditional DMARDs. Our data show a similar tendency since we found pericarditis in 20 % of patients receiving biologic agents and in 12 % of patients receiving traditional DMARDs, thus supporting the suggestion that pericarditis in AOSD patients should be considered a red flag for clinicians [15].
In the Chinese cohort, a higher prevalence of lymphadenopathy, hepatomegaly, maculopapular rash and pharyngitis was noted. In our series, rash, lymphadenopathy and sore throat were sensitive (>60 %) and specific (>60 %), suggesting that these 3 features are useful for establishing a diagnosis of AOSD in the Italian cohort.
Hemophagocytic syndrome is a rare life-threatening syndrome that can complicate the course of AOSD. It is suggested that prevalence of RHS in AOSD is underestimated due to underdiagnosis in several cases. In our cohort, we observed RHS only in 2.8 % of patients. This number is low in comparison to other AOSD series, however is consistent with the recently reported results of Zhang Y. et al. who retrospectively identified 10 cases of RHS among 315 AOSD patients (3.2 %) [16].
With regard to clinical course, monocyclic subset was reported most commonly [14, 17], whereas in our cohort, most frequent subsets were polycyclic systemic pattern (40.8 %) and chronic articular polyciclic (30.7 %).
Particular attention should be paid to the laboratory values, since it may be a very helpful diagnostic hint for AOSD. Among other laboratory features, the white blood cell count seems to be an interesting predictor of patient outcome. Increased white blood cell counts were associated with AOSD relapses, whereas other studies showed a significant association between elevated serum ESR or CRP and a poor prognoses or higher relapse rates [9, 18].
Notably, the Italian cases experienced hyperferritinemia and showed a remarkably higher level of serum ferritin with respect to Turkey and Chinese series. Besides total ferritin level, the diagnostic interest of the glycosylated ferritin (GF) has been suggested [19]. Unfortunately, GF determination was not available in Italian routine clinical practice at the time of the study enrollment.
The determination of RF and ANA is also recommended, since it can be effective in narrowing the differential diagnosis. The frequency of RF and ANA positivity, always at low titre in our patients, is comparable to other studies.
The treatment of AOSD remains largely empirical, based on few case studies but not on randomized trials. NSAIDs are traditionally recommended as initial treatment of AOSD, although they are rarely effective in controlling the disease with a response rate of 20–25 % [20]. Corticosteroids are usually required to induce symptom remission and are indicated in case of life-threatening complications in AOSD [21, 22]. To reduce the effects of the steroids, immunomodulatory agents as methotrexate (MTX) can be used as second-line treatment [23]. However, if conventional immunosuppressive therapy is not effective, biological agents targeting IL-1, IL-6 and TNF-α represent major therapeutic advances. In our cases, corticosteroids and traditional DMARDs have not been able to control the disease in 21.9 % of patients while biologics have been shown positive effects. The efficacy of infliximab (anti-TNF-α) in corticosteroid- and MTX-resistant AOSD patients was demonstrated in several case series and case reports [24, 25]. Infliximab is generally well tolerated; however, it has been associated with side effects including infusion reactions, skin rash, infections, fulminant hepatitis and exacerbation of heart failure. Switching from one TNF-α inhibitor to another may be useful. In fact, etanercept (anti-TNF-α) was used successfully for the treatment of AOSD patients with complications, whereas adalimumab was effective as second-line biologic drug in etanercept or infliximab not responders [26]. Treatment with IL-1 inhibitor (anakinra) resulted in rapid and complete resolution of both systemic and articular manifestations as well as normalization of inflammatory marker levels [27]. Three of our cases received anti-IL-6 receptor, which has been considered for treating AOSD. A recent review of 35 patients reported that 86 % of tocilizumab-treated patients experienced prompt articular improvement, and 96 % experienced a disappearance of systemic symptoms [28].
We conducted a nationwide survey of AOSD, which provides important information on the clinical, laboratory and therapeutic features in the greatest cohort of Italian patients. Nevertheless, the present study has few limitations. First, the study was retrospective and had to contend with some degree of missing clinical and laboratory investigation data. Therefore, we await additional studies from other institutions in different countries to enrich and confirm the present results.
References
Still GF (1897) On a form of chronic joint disease in children. Med Chir Trans 80:47–60
Bywaters EG (1971) Still’s disease in the adult. Ann Rheum Dis 30:121
Magadur-Joly G, Billaud E, Barrier JH et al (1995) Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis 54:587–590
Cagatay Y, Gul A, Cagatay A et al (2009) Adult-onset Still’s disease. Int J Clin Pract 63:1050–1055
Fautrel B, Zing E, Golmard J-L et al (2002) Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore) 81:194–200
Yamaguchi M, Ohta A, Tsunematsu T et al (1992) Preliminary criteria for classification of adult Still’s disease. J Rheumatol 19:424–430
Efthimiou P, Paik PK, Bielory L (2006) Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis 65:564–572
Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P (2014) Adult-onset Still’s disease. Autoimmun Rev 13:708–722
Kong X-D, Xu D, Zhang W, Zhao Y, Zeng X, Zhang F (2010) Clinical features and prognosis in adult-onset Still’s disease: a study of 104 cases. Clin Rheumatol 29:1015–1019
Asanuma YF, Mimura T, Tsuboi H et al (2015) Nationwide epidemiological survey of 169 patients with adult Still’s disease in Japan. Mod Rheumatol 25:393–400
Scirè CA, Cavagna L, Perotti C, Bruschi E, Caporali R, Montecucco C (2006) Diagnostic value of procalcitonin measurement in febrile patients with systemic autoimmune diseases. Clin Exp Rheumatol 24:123–128
Priori R, Ceccarelli F, Barone F, Iagnocco A, Valesini G (2008) Clinical, biological and sonographic response to IL-1 blockade in adult-onset Still’s disease. Clin Exp Rheumatol 26:933–937
Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG (2010) Adult onset Still’s disease: clinical presentation in a large cohort of Italian patients. Clin Exp Rheumatol 28:41–48
Colina M, Zucchini W, Ciancio G, Orzincolo C, Trotta F, Govoni M (2011) The evolution of adult-onset Still disease: an observational and comparative study in a cohort of 76 Italian patients. Semin Arthritis Rheum 41:279–285
Dall’Ara F, Frassi M, Tincani A, Airò P. A retrospective study of patients with adult-onset Still’s disease: is pericarditis a possible predictor for biological disease-modifying anti-rheumatic drugs need? Clin Rheumatol. 2016. [Epub ahead of print]
Zhang Y, Yang Y, Bai Y, Yang D, Xiong Y, Zeng X. Clinical characteristics and follow-up analysis of adult-onset Still’s disease complicated by hemophagocytic lymphohistiocytosis. Clin Rheumatol. 2016. [Epub ahead of print]
Cush JJ, Medsger TAJR, Christy WC, Herbert DC, Cooperstein LA (1987) Adult-onset Still’s disease. Clinical course and outcome. Arthritis Rheum 30:186–194
Kim H-A, Sung J-M, Suh C-H (2012) Therapeutic responses and prognosis in adult-onset Still’s disease. Rheumatol Int 32:1291–1298
Fautrel B, Le Moël G, Saint-Marcoux B et al (2001) Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol 28:322–329
Fautrel B (2008) Adult onset Still disease. Best Pract Res Clin Rheumatol 22:773–792
Reginato AJ, Schumacher HR Jr, Baker DG et al (1987) Adult onset Still’s disease: experience in 23 patients and literature review with emphasis on organ failure. Semin Arthritis Rheum 17:39–57
Bisagni-Faure A, Job-Deslandre C, Menkes CJ (1992) Intravenous methylprednisolone pulse therapy in Still’s disease. J Rheumatol 19:1487–1488
Fautrel B, Borget C, Rozenberg S et al (1999) Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol 26:373–378
Dilhuydy MS, Vatan R, Etienne G, Longy-Boursier M, Mercié P (2005) Prolonged efficacy of infliximab for refractory adult-onset Still’s disease. Clin Exp Rheumatol 23:121–122
Olivieri I, de Stefano G, Padula A, La Gala A, de Stefano C (2003) Infliximab in a case of early adult-onset Still’s disease. Clin Rheumatol 22:369–370
Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG (2010) Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum 62:2530–2535
Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis S-NC (2007) Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann Rheum Dis 66:842–843
de Boysson H, Fevrier J, Nicolle A, Auzary C, Geffray L (2013) Tocilizumab in the treatment of the adult-onset Still’s disease: current clinical evidence. Clin Rheumatol 32:141–147
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This retrospective study was approved by Padua Hospital Ethics Committee.
Disclosures
None.
Rights and permissions
About this article

Cite this article
Sfriso, P., Priori, R., Valesini, G. et al. Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol 35, 1683–1689 (2016). https://doi.org/10.1007/s10067-016-3308-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-016-3308-8