
Abstract
Rhabdomyosarcoma is the most common soft-tissue sarcoma affecting children and adolescents. It is defined as a malignant neoplasm characterized by morphologic, immunohistochemical, ultrastructural, or molecular genetic evidence of primary skeletal muscle differentiation, usually in the absence of any other pattern of differentiation. Primary intracranial rhabdomyosarcoma (PIRMS) is an extremely rare neoplasm, with only 60 cases reported in the literature, and generally has poor prognosis with an overall survival of only 9.1 months. The DICER1 gene encodes an RNA endoribonuclease that plays a key role in gene expression regulation through the production of small RNAs. Herein, we report two cases of PIRMS with somatic DICER1 mutation showing morphological and immunohistochemical evidence of primary skeletal muscle differentiation; the two cases share common clinical features, including young age, supratentorial tumor, and onset of intratumoral bleeding. Although methylation profiling was not performed, both cases shared clinical and pathological characteristics in common with recently proposed methylation entity “spindle cell sarcoma with rhabdomyosarcoma-like features, DICER1 mutant (SCS-RMSlike-DICER1)’’. Our cases provide further evidence of the link between primary intracranial sarcoma and DICER1 mutation which may form a distinct entity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhabdomyosarcoma is defined as a malignant neoplasm that shows morphologic, immunohistochemical, ultrastructural, or molecular genetic evidence of primary skeletal muscle differentiation, usually in the absence of any other pattern of differentiation. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and adolescents and is classified into two major subtypes, namely, embryonal and alveolar, based on distinct clinicopathological features, genetic alterations, and prognosis. The most common site of involvement is the head and neck region (~ 40%; including the nasal cavity, parameninges, and orbit), followed by the urogenital tract (~ 25%; most commonly in the paratesticular region in adolescents), and the extremities (~ 20%). Primary intracranial rhabdomyosarcoma (PIRMS) is an extremely rare neoplasm, with only 60 cases reported in the literature, and generally has poor prognosis, with an overall survival of only 9.1 months [1]. However, several recent studies have reported prolonged survival with the use of multi-modality treatment [1,2,3].
The DICER1 gene, which is located at 14q32.13, encodes an RNA endoribonuclease that plays a key role in gene expression regulation through the production of small RNAs [4]. DICER1 syndrome (OMIM: 601200) is a rare genetic disorder that predisposes individuals to the development of tumors, both benign and malignant, including pleuropulmonary blastoma, cystic nephroma, multinodular goiter, ovarian Sertoli-Leydig cell tumor, embryonal rhabdomyosarcoma, and other rare tumor entities. This disease primarily affects individuals in the pediatric and adolescent age range. The two-hit hypothesis based on the second somatic mutations in the RNase IIIb domain of the DICER1 gene has been proposed as a mechanism responsible for causing rare tumors [5].
Genetically, alveolar rhabdomyosarcoma usually carries specific chromosomal translocations that produce PAX3- or PAX7-FOXO1 fusion genes, whereas embryonal rhabdomyosarcoma is commonly characterized by loss of heterozygosity at 11p15.5 and gains of chromosomes 2, 8, and 12 in varying combinations [6, 7]. The DICER1 mutation in rhabdomyosarcoma is reported in embryonal rhabdomyosarcoma and originates within the urogenital tract, especially the uterine cervix, which is an uncommon site for rhabdomyosarcoma [8,9,10,11,12,13,14]. Histological features of rhabdomyosarcoma of the uterine cervix included cartilaginous nodules in nearly half of the cases [15].
Herein, we reported two cases of supratentorial PIRMS with somatic DICER1 mutation that caused intracerebral hematoma in a 10-year-old girl (patient #1) and a 29-year-old male (patient #2). The former case has followed a long-term course of 5 years.
Clinical summary
Patient #1 is a 10-year-old girl who was referred to the hospital because of sudden onset of severe headache and upper extremity paralysis. She experienced vomiting and dressing apraxia 2 months prior and had surgery for evacuation of hematoma in the right parietal lobe. Upon admission to our institution, cranial CT and magnetic resonance imaging (MRI) revealed a hematoma in the same area, measuring 6.0 cm × 6.0 cm × 6.0 cm, with perifocal edema (Fig. 1a, b, c). A neoplasm was identified during the second operation for evacuation of hematoma, and the initial pathologic diagnosis was malignant tumor, NOS. Whole-body CT scan, FDG-PET scan, and bone marrow biopsy results were normal. She was administered with vincristine and focal radiotherapy on suspicion of primitive neuroectodermal tumor; treatment was later changed to a stronger chemotherapeutic regimen. Recurrence occurred 2 months later, and tumor excision was performed. Combined radiotherapy (50.4 Gy) and multi-agent chemotherapy comprising doxorubicin, etoposide, and cisplatin were given based on the final diagnosis of rhabdomyosarcoma and the enhanced lesion in the excisional cavity on MRI. Positive therapeutic response was observed for 41 months until the second recurrence. The tumor was incompletely excised during the subsequent recurrence because of infiltration of a large vein; therefore, gamma knife therapy was initiated. Disease recurrence adjacent to the resection cavity was observed on follow-up imaging at 16 months after the second tumor resection. The third tumor resection was recently performed and confirmed the recurrence of rhabdomyosarcoma. At 68 months from the initial therapy, the patient is alive under treatment.

Radiological images of intracranial rhabdomyosarcoma; patient #1 (a–c) and patient #2 (d–f). The hematoma in the right parietal lobe (measuring 6.0 cm × 6.0 cm × 6.0 cm) with edema was revealed on CT (a), and was observed to be iso-intense on the T1-weighted image (b) and hyper-intense on the T2-weighted image (c), suggesting acute stage of hematoma. A left parietal lobe mass with a diameter of 7.5 cm (d). MRI revealed that the mass was hypo-intense on the T1-weighted image (e) and the hyper-intense on T2-weighted image (f), suggesting acute stage of hematoma
Patient #2 is a 29-year-old male who presented right-sided hemiparesis. Brain CT scan showed a 7.5 cm left parietal lobe mass with internal bleeding that causes ventricular rupture (Fig. 1d). MRI revealed that the mass appeared hypo-intense on T1-weighted images and hyper-intense on T2-weighted images (Fig. 1e, f), and heterogeneously enhancement on contrast enhanced using gadolinium. The tumor was removed completely by craniotomy. The patient received chemoradiotherapy with vincristine, focal radiation of 32.4 Gy, and craniospinal radiation of 23.4 Gy. Afterwards, the patient underwent VAC chemotherapy comprising vincristine, actinomycin D, and cyclophosphamide, and alive with no evidence of recurrence at 11 months after disease onset.
Pathological findings
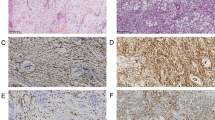
Microscopic features of the two cases were similar; results showed sarcomatous neoplasms that were highly cellular with brisk mitotic activity and admixed with large pools of hemorrhage. The tumor cells were composed of diffuse proliferation of immature spindle and ovoid cells that were arranged in fascicular and storiform patterns (Fig. 2a, h). Individual cells exhibited marked cytologic atypia with pleomorphic nuclei, and multi-nucleated giant cells were often observed (Fig. 2b, j). Focal clusters and scattered rhabdomyoblasts, characterized by large round or oval eosinophilic cells with eccentric nuclei and abundant eosinophilic granular cytoplasm, were also noted (Fig. 2c, i). Patient #1 showed tumor cells with eosinophilic cytoplastic globules near the site of hemorrhage (Fig. 2d). Patient #1 showed myxoid area and a small focus of cartilaginous differentiation (Fig. 2e). Reticulin staining revealed abundant intercellular basement membrane deposition. Immunohistochemistry results showed that the tumor cells were diffusely and strongly positive for vimentin. Focal positivity for muscle markers, including desmin (Fig. 2f, k), HHF-35, myogenin (Fig. 2g, l), and myoglobin, were apparent in tumor cells with eosinophilic cytoplasm. Myoglobin staining was highly specific to rhabdomyoblast. Immunoreactivity for α-SMA varied; signals were scattered in patient #1 and were present in 30% of tumor cells in patient #2. INI1 expression was retained. Staining for S-100 protein, GFAP, synaptophysin, EMA was negative. In both cases, the sarcomas showed differentiation from undifferentiated cells to rhabdomyoblast; therefore, the tumors were histologically diagnosed as rhabdomyosarcoma.

Microscopic and immunohistochemical analyses of intracranial rhabdomyosarcoma; patient #1 (a–g) and patient #2 (h–l). Diffuse proliferation of immature spindle cells arranged in a fascicular pattern (a, h). At high magnification, the tumor cells exhibited pleomorphic nuclei and eosinophilic cytoplasm (b, j). Frequent mitosis and scattered multi-nucleated giant cells were observed. Focal clusters of rhabdomyoblasts characterized by eccentric nuclei and abundant eosinophilic granular cytoplasm were noted (c, i). Patient #1 showed tumor cells with eosinophilic cytoplastic globules (d) and a small focus of cartilaginous differentiation (e). Focal positivity for desmin in the cytoplasm of tumor cells (f, k). Focal positivity for myogenin in the nuclei of tumor cells (g, l). DICER1 RNase IIIb mutations in PIRMS (m). c.5425G>A, p.G1809R somatic mutation in patient #1. The same mutation was detected during both onset and recurrence. The c.5127T>A, p.D1709E mutation in patient #2
Molecular analysis
Somatic DICER1 and TP53 mutations
DNA was extracted from fresh frozen tumor and non-tumor brain tissues in patient #1 and formalin-fixed and paraffin-embedded (FFPE) tumor tissues in patient #2 using DNeasy Blood and Tissue Kit (Qiagen, Tokyo, Japan) and GeneRead DNA FFPE Kit (Qiagen, Tokyo, Japan), respectively. Sanger sequencing of the RNase IIIb domain of DICER1 was performed using the following primer sets: forward, 5′-CCCCTCAGATTGTTACCAGC-3′; reverse, 5′-CGTTTTGAACAGCACTAACCTC-3′ and forward, 5′-TCTGAGGAGGATGAAGAGAAAG-3′; reverse 5′-CGTTTTGAACAGCACTAACCTC -3′. As for tumor, DNA extracted from patient #l target sequencing of all coding exons of the 93 selected brain tumor-related genes (Supplementary Table 1) was performed as previously described [16].
Non-synonymous mutations in the DICER1 RNase IIIb domain were detected in both tumor samples. These mutations included c.5425G>A, p.G1809R in patient #1 and c.5127T>A, p.D1709E in patient #2 (Fig. 2m). In patient #1, no variant was detected in all coding exons of DICER1 in non-tumor control tissues. In patient #2, only tumor DNA was available, while the non-tumor control sample was not available. The somatic mutation c.569C>T, p.P190L in TP53 was also detected in patient #1.
Discussion
Recently, Koelsche et al. proposed a new entity of intracranial sarcoma, “spindle cell sarcoma with rhabdomyosarcoma-like features, DICER1 mutant (SCS-RMSlike-DICER1)’’ based on the result of methylation analysis of 22 primary intracranial sarcoma cases [17]. As represented by the nomenclature, histologically rhabdomyoblasts or rhabdomyoblast-like cells were observed in all cases and most tumor cells were spindle shape, although supplier diagnosis of these 22 cases were variable including sarcoma NOS (n = 15), malignant tumor NOS (n = 2), embryonal rhabdomyosarcoma (n = 1), gliosarcoma (n = 1), glioblastoma (n = 1), mesenchymal chondrosarcoma (n = 1), and PNET (n = 1) [17]. DICER1 hot spot mutations were detected in 21/22 cases and TP53 mutations in 12/22 cases [17]. Following this report, however, Lee et al. presented another cases of primary intracranial sarcoma which display the same molecular characteristics as “SCS-RMSlike-DICER1″ but different morphological characteristics: they harbor DICER1 mutation and “SCS-RMSlike-DICER1” methylation pattern, but were morphologically characterized as pleomorphic rather than predominantly spindled or round cell with myogenic differentiation [18]. Accordingly, the authors proposed a broader term “Primary intracranial sarcoma, DICER1-mutant”. Thus, the definition of the newly proposed entity of intracranial sarcomas with DICER1 mutation, whether or not these tumors truly form a single entity despite the histological diversity, remains controversial.
In the present study, we reported two cases of supratentorial PIRMS harboring DICER1 mutation. In both cases, the tumors were characterized by immature spindle and ovoid cells with polymorphism and multi-nucleated giant cells, and showed clear skeletal muscle differentiation based on morphological and immunohistochemical analyses. Differential diagnosis, including other brain tumors that occasionally showed skeletal muscle components, such as medulloblastoma, atypical teratoid/rhabdoid tumor, gliosarcoma, anaplastic meningioma, and germ cell tumor, were excluded from the site and the immunohistochemistry results. The tumor samples from the present two cases harbored somatic hot spot mutations within the DICER1 RNase IIIb domain, namely, p.G1809R (c.5425G>A) in patient #1 and p.D1709E (c.5127T>A) in patient #2; these mutations were previously described as somatic mutations in intracranial sarcoma, pleuropulmonary blastoma, cystic nephroma, Sertoli–Leydig cell tumors, rhabdomyosarcoma of the ovary, and other DICER1-related neoplasms (Table 1) [4, 10, 12, 17, 19]. For patient #2, only tumor tissue was available without a paired normal sample; thus, the somatic versus germ line status of the identified DICER1 mutation could not be determined. On the other hand, the mutation in patient #1 was confirmed to be somatic. As recognized in previous reports, primary intracranial sarcomas with DICER1 mutation showed a diverse histologic spectrum, ranging from cases presenting immature cells to cases with distinct differentiation into skeletal muscle; latter cases were similar to the features observed in our two cases.
The clinical features and behaviors of intracranial sarcoma harboring DICER1 mutations have not been previously investigated because of limited cases and follow-up sessions that are insufficient to derive reliable conclusions. In the present study, both cases were young and showed supratentorial tumors causing intracerebral hematoma. In 28 cases of intracranial sarcoma with DICER1 mutations, including our two cases, the age distribution ranged from 0 to 76 years with a median age of 13 years, and the gender distribution was almost equal between females (n = 15) and males (n = 13) [17, 18, 20, 21]. A total of 26 cases had supratentorial tumor locations, except for two cases, in which the tumors were located in the cerebellopontine angle and cerebellum. Intratumoral hemorrhage was observed in all six cases in which image finding were mentioned, and it is considered that there is a feature easy to accompany bleeding. Previous studies have reported that PIRMS is commonly seen in infants and young children and the most common site of involvement in children is the cerebellum unlike adults in whom a supratentorial predilection [22,23,24]. In 60 cases of PIRMS, the age distribution ranged from 0 to 68 years with a median age of 18 years; 70% of cases were less than 18 years old, and M:F ratio was 1.3:1. The tumor located in supratentorial in 32, infratentorial in 20, supra-infratentorial in 3, and pineal are in 5 [1,2,3, 17, 22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. Intracranial hemorrhage, not usually seen in CNS sarcoma, was also previously described in PIRMS [1, 22, 26, 27, 36]. PIRMS has poor prognosis, with an overall survival of only 9.1 months; however, several recent studies reported prolonged survival with the use of multi-modality treatment [1,2,3]. The two patients in this study multimodal treatment, including surgery, chemotherapy, and radiation, well worked.
In summary, we reported two cases of PIRMS harboring DICER1 mutations. Although methylation profiling was not performed, both cases shared clinical and pathological characteristics in common with recently proposed methylation entity “SCS-RMSlike-DICER1” [17]. Our cases provide further evidence of the link between primary intracranial sarcoma and DICER1 mutation which may form a distinct entity.
References
Guilcher GM, Hendson G, Goddard K et al (2008) Successful treatment of a child with a primary intracranial rhabdomyosarcoma with chemotherapy and radiation therapy. J Neurooncol 86:79–82
Al-Gahtany M, Shroff M, Bouffet E et al (2003) Primary central nervous system sarcomas in children: clinical, radiological, and pathological features. Childs Nerv Syst 19:808–817
Ishi Y, Yamaguchi S, Iguchi A et al (2016) Primary pineal rhabdomyosarcoma successfully treated by high-dose chemotherapy followed by autologous peripheral blood stem cell transplantation: case report. J Neurosurg Pediatr 18:41–45
Foulkes WD, Priest JR, Duchaine TF (2014) DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14:662–672
Robertson JC, Jorcyk CL, Oxford JT (2018) DICER1 Syndrome: DICER1 mutations in rare cancers. Cancers (Basel). https://doi.org/10.3390/cancers10050143
Liu C, Li D, Jiang J et al (2014) Analysis of molecular cytogenetic alteration in rhabdomyosarcoma by array comparative genomic hybridization. PLoS One 9:e94924
Nishimura R, Takita J, Sato-Otsubo A et al (2013) Characterization of genetic lesions in rhabdomyosarcoma using a high-density single nucleotide polymorphism array. Cancer Sci 104:856–864
Doros L, Yang J, Dehner L et al (2012) DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 59:558–560
Foulkes WD, Bahubeshi A, Hamel N et al (2011) Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32:1381–1384
Heravi-Moussavi A, Anglesio MS, Cheng S-WG et al (2012) Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 366:234–242
de Kock L, Boshari T, Martinelli F et al (2016) Adult-onset cervical embryonal rhabdomyosarcoma and DICER1 mutations. J Low Genit Tract Dis 20:e8–e10
de Kock L, Druker H, Weber E et al (2015) Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol 46:917–922
de Kock L, Rivera B, Revil T et al (2017) Sequencing of DICER1 in sarcomas identifies biallelic somatic DICER1 mutations in an adult-onset embryonal rhabdomyosarcoma. Br J Cancer 116:1621–1626
Tomiak E, de Kock L, Grynspan D et al (2014) DICER1 mutations in an adolescent with cervical embryonal rhabdomyosarcoma (cERMS). Pediatr Blood Cancer 61:568–569
Dehner LP, Jarzembowski JA, Hill DA (2012) Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25:602–614
Nakano Y, Tomiyama A, Kohno T et al (2019) Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 36:14–19
Koelsche C, Mynarek M, Schrimpf D et al (2018) Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol 136:327–337
Lee JC, Villanueva-Meyer JE, Ferris SP et al (2019) Primary intracranial sarcomas with DICER1 mutation often contain prominent eosinophilic cytoplasmic globules and can occur in the setting of neurofibromatosis type 1. Acta Neuropathol 137:521–525
Bean GR, Anderson J, Sangoi AR et al (2019) DICER1 mutations are frequent in mullerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol 32:280–289
Das A, Roy P, Modi SK et al (2019) Germline DICER1-mutant intracranial sarcoma with dual chondroid and spindle cell morphology and pulmonary metastases treated with multimodal therapy. Pediatr Blood Cancer 66:e27744
de Kock L, Geoffrion D, Rivera B et al (2018) Multiple DICER1-related tumors in a child with a large interstitial 14q32 deletion. Genes Chromosomes Cancer 57:223–230
Dropcho EJ, Allen JC (1987) Primary intracranial rhabdomyosa: case report and review of the literature. J Neurooncol 5:139–150
Celli P, Cervoni L, Maraglino C (1998) Primary rhabdomyosarcoma of the brain: observation on a case with clinical and radiological evidence of cure. J Neurooncol 36:259–267
Pirillo V, Cipriano Cecchi P, Tripodi M et al (2011) Primary cerebral alveolar rhabdomyosarcoma in adult. Rare Tumors 3:e26
Hawkins C, Muller P, Bilbao JM (1999) April 1999–44 year old man with a bleeding intracerebral tumor. Brain Pathol 9:741–742
Mitsuhashi T, Mori K, Wada R et al (2002) Primary rhabdomyosarcoma associated with tumoral hemorrhage–case report. Neuro Med Chir (Tokyo) 42:73–77
Grebe HP, Steube D (2008) Primary cerebral rhabdomyosarcoma presenting as haemorrhagic stroke. Zentralbl Neurochir 69:93–95
Khalatbari MR, Hamidi M, Moharamzad Y (2013) Primary alveolar rhabdomyosarcoma of the brain with long-term survival. J Neurooncol 115:131–133
Benesch M, von Bueren AO, Dantonello T et al (2013) Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111:337–345
Caporlingua F, Lapadula G, Antonelli M et al (2014) Pleomorphic rhabdomyosarcoma of the cerebellopontine angle in an adult: a review of literature. BMJ Case Rep. https://doi.org/10.1136/bcr-2013-203257
Lau SK, Cykowski MD, Desai S et al (2015) Primary rhabdomyosarcoma of the pineal gland. Am J Clin Pathol 143:728–733
Maher OM, Khatua S, Mukherjee D et al (2016) Primary intracranial soft tissue sarcomas in children, adolescents, and young adults: single institution experience and review of the literature. J Neurooncol 127:155–163
Nair P, Das KK, Srivastava AK et al (2017) Primary intracranial rhabdomyosarcoma of the cerebellopontine angle mimicking a vestibular schwannoma in a child. Asian J Neurosurg 12:109–111
Yoshida K, Miwa T, Akiyama T et al (2018) Primary intracranial rhabdomyosarcoma in the cerebellopontine angle resected after preoperative embolization. World Neurosurg 116:110–115
Desai KB, Mella D, Pan E (2019) An adult patient with rare primary intracranial alveolar rhabdomyosarcoma. Anticancer Res 39:3067–3070
Tomei G, Grimoldi N, Cappricci E et al (1989) Primary intracranial rhabdomyosarcoma: report of two cases. Childs Nerv Syst 5:246–249
Acknowledgements
We thank the advice on pathological diagnosis by Dr. Takanori Hirose (Department of Pathology, Kobe University; patient #1) and Dr. Junko Hirato (Department of Pathology, Gunma University; patient #2), and technical assistance by Ms. Erika Komura (Kanazawa University) and Ms. Yuko Hibiya (National Cancer Center Research Institute).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
List of 93 sequenced genes (DOCX 15 kb)
Rights and permissions
About this article

Cite this article
Sakaguchi, M., Nakano, Y., Honda-Kitahara, M. et al. Two cases of primary supratentorial intracranial rhabdomyosarcoma with DICER1 mutation which may belong to a “spindle cell sarcoma with rhabdomyosarcoma-like feature, DICER1 mutant”. Brain Tumor Pathol 36, 174–182 (2019). https://doi.org/10.1007/s10014-019-00352-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-019-00352-z