
Abstract
Electrochemical impedance spectroscopy (EIS) and voltammetry of immobilized particles (VIMP) measurements using air-saturated mineral water and 0.10 M NaClO4 aqueous solution as electrolytes were applied to eurocent coins and a set of copper/bronze coins from the late nineteenth century exhibiting significant heterogeneity in their degree and type of corrosion. The obtained data presented satisfactory repeatability being fitted to relatively simple equivalent circuits which were dependent on the electrolyte and bias potential, the more satisfactory conditions being obtained using the reduction of dissolved oxygen as a redox probe. Consistent data were obtained using VIMP and EIS characterizing different corrosion patterns, and establishing the possibility of discriminating different monetary emissions in favorable cases of high level of corrosion.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-destructive and non-invasive techniques are in general required for the analysis of cultural heritage samples as the investigations on the materials have to respect several limitations mainly due to the unicity of the piece. Moreover, in the case of metal artefacts, the corrosion processes and the studies about the patina formation have to be considered. Although it is not possible to stop the corrosion process, it is possible to monitor it and characterize the products formed on the surface of the metals. Accordingly, the patina has been subject of several scientific studies because the formation of a noble one can help the conservation of the metal without (or minimal) additional intervention, as demanded by the philosophy of the modern conservation [1,2,3,4,5,6,7]. However, the surface of a metal is presented as a heterogeneous multi-layer structure where it is often possible to distinguish a primary, a secondary and a tertiary patinas [8, 9].
There are several techniques, optical and electron microscopies, X-ray diffraction, and different spectroscopies, applicable to the characterization of the patina [10,11,12,13], but electrochemical techniques have contributed to a better understanding the evolution of corrosion processes on ancient metals [5, 7, 14, 15] as well as they have been shown to be useful into the archaeometric field [8, 16,17,18,19,20]. In particular, electrochemical impedance spectroscopy (EIS), which is a consolidated technique for studying corrosion phenomena and coating performances on modern materials [21,22,23,24,25,26,27], is being increasingly applied to the study of archaeological samples [5, 7,8,9, 16, 28,29,30,31,32,33,34,35,36].
In general, impedance spectra are recorded at the open circuit potential (OCP) with the aid of a redox probe which behaves reversibly, typically the Fe (CN)63−/4− couple. The use of this probe as well as aggressive electrolytes (acidic, alkaline and/or chloride-containing), usual in studies on protective coatings of metal artefacts, is obviously hindered for studying archaeological objects and works of art. In order to apply EIS for studying archaeological objects, several difficulties have to be accounted: i) the need of using operation conditions able to yield archaeometric information without damage to the pieces; ii) the area normalization commonly used in studies on protective coatings cannot be directly applied here because the objects frequently present irregular and porous and/or anfractuous surfaces. Additionally, it is pertinent to note that the system should operate under steady state conditions accomplishing the demands of the linear systems' theory [37, 38]; i.e. the system should be time-invariant during the acquisition of the impedance data satisfying the Kramers-Kronig (K-K) transfer functions [39, 40]. Under these conditions, EIS allows monitoring the resistance, porosity, roughness of the patina and/or protective coatings on metallic surfaces.
One way of application of EIS for archaeometric purposes consists of the determination of the impedance response of the archaeological object used as a working electrode in contact with a suitable electrolyte. In previous works, we used an electrochemical cell adapted for testing coins and other metallic objects [31] for discriminating monetary emissions of relatively high homogeneity [8, 16], incorporating air-saturated mineral water, recommended ensuring the morphological, structural and chemical integrity of the archaeological objects [41], and using the reduction of dissolved oxygen a redox probe [8, 16]. As far as this last condition implies the application of a bias potential in general separated from the OCP, and that potential (ca. −0.6 V vs. Ag/AgCl) makes possible the electrochemical reduction of some corrosion products, the system operates, strictly, under non-stationary conditions, a situation used to study electrochemical reaction pathways [42, 43].
The current report presents a study aimed to: i) investigate the suitability of impedance measurements for characterizing archaeological metals, and, ii) optimizing the experimental conditions (electrolyte, bias potential) in order to study archaeological metal objects having large heterogeneity in their ‘corrosion history’. As far as the maintenance of the integrity of the object and the electrochemical operation conditions during EIS experiments are essential requirements, we have used the voltammetry of immobilized particles methodology (VIMP) for testing the possible alteration of the patina. The VIMP is a solid-state electrochemical technique developed by Scholz et al. that permits the performance of localized analyses using amounts of sample at the sub-microgram level [44, 45]. Due to its inherently high sensitivity, the VIMP is particularly useful in the field of cultural heritage [46, 47] and has been extensively used to study archaeological metal patinas [8, 16, 48]. In this study, VIMP and EIS have been used to characterize the impedance response of two series of copper/bronze coins, contemporary eurocents and a heterogeneous set of Spanish chavos produced in 1885–1895, using air-saturated mineral water and 0.10 M NaClO4 as electrolytes.
Experimental
Samples
A series of six Spanish 5-eurocent coins and a set of bronze coins produced at the late nineteenth century and beginning of the twentieth century were studied. These were as follows: C1, C3, C5: 10 cts. Alphonse XII, Spain, 1877; C2: 10 cts. French Republic, 1912; C4 and C6: 10 cts. Spanish Republic, 1870. The eurocents were selected as showing a uniform, smooth corrosion characterized by a brownish patina. The nineteenth century coins exhibited quite different condition of conservation, from a brownish uniform surface to a black-greenish patina presenting locally deposits of green-powdered corrosion products. The whole group of analyzed coins is shown in Fig. 1.

The set of the studied ancient bronze coins showing a different degree of surface deterioration
Electrochemical instrumentation and procedures
All electrochemical experiments were carried out using a three-electrode arrangement connected to a CH 660c potentiostat with a Pt mesh auxiliary electrode and an Ag/AgCl (3 M NaCl) reference electrode. VIMP experiments were performed upon abrasive transference of sub-microsamples from the corrosion patina of the coins to paraffin-impregnated commercial graphite bars (Alpino CH type, surface area of 0.0314 cm2) using air-saturated 0.25 M HAc/NaAc aqueous acetate buffer at pH 4.75. No deaeration was performed in order to mimic the conditions usable in ‘in-field’ analysis of cultural heritage. For each coin, three to five replicate measurements were performed sampling on different spots of the coin surface. The sample-modified graphite electrodes were placed in the electrochemical cell so that only the lower electrode end came into contact with the electrolyte solution and then the voltammetric measurements were carried out. Square wave voltammograms (SWVs) were recorded in both negative- and positive-going potential scans using potential step increments between 4 and 10 mV, square wave amplitudes between 25 and 100 mV and frequencies between 2 and 500 Hz.
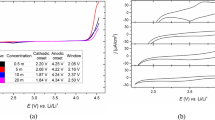
EIS experiments were performed in the same electrolytes using a modified cell, adapted for partial immersion of metallic objects, already described [8, 16, 31]. Spectra were recorded in the 0.1 and 100,000 Hz frequency range with amplitude of 5 mV at different bias potentials: 0.2, 0.0, −0.2, −0.4, −0.6, and − 0.8 V vs. Ag/AgCl after 2 min of equilibration time. The time for recording each spectrum was of 5 min. For each coin, three-five replicate experiments were performed varying the position of the clamp and the immersed region but maintaining an immersed area of ca. 1 cm2. As electrolytes, air-saturated mineral water from Bejís (Comunitat Valenciana, Spain) and sodium perchlorate (Merck) 0.10 M have been used. The chemical composition of the mineral water is dry residual (159 mg L−1, HCO3−: 163 mg L−1, SO42−: 16 mg L−1, Cl−: 1.2 mg L−1, SiO2: 4.3 mg L−1, Ca2+: 47 mg L−1, Mg2+: 6.2 mg L-1, Na+: 2.8 mg L−1). This composition is taken as reported from the water producer as not being measured in the present study.
Results and discussion
VIMP
Figure 2 compares the negative-going potential scan square wave voltammograms for sub-microsamples from the corrosion layers of a) eurocent coin (three replicate experiments), b) coin C1, c) coin C3 and d) coin C4, attached to graphite electrode in contact with air-saturated 0.25 M aqueous acetate buffer at pH 4.75. The selected conditions resulting from compromise between repeatability and peak resolution were potential step increment 4 mV, square wave amplitude 25 mV, frequency 5 Hz. In the case of coins C3–C6, the sampling was performed in brown-black areas of the coins, by passing regions were deposits of green corrosion products were accumulated. The voltammetric response, in agreement with that described in previous reports [8, 16], consists of a main cathodic signal at −0.10 V vs. Ag/AgCl (I) followed by a more or less defined shoulder at ca. −0.45 V (II) which precedes the prominent rising current due to the hydrogen evolution reaction (HER). The signal I corresponds to the reduction of cuprite (Cu2O) and other accompanying copper corrosion products (azurite, malachite, brochantite, atacamite) whereas the signal II can be attributed to the reduction of tenorite (CuO). In agreement with the expectances, the ratio between the intensity of the signals II and I, I(II)/I(I), measured using the baseline depicted in Fig. 2b–d, increases on increasing the degree of alteration of the coin. This ratio can be considered as representative of the averaged tenorite/cuprite ratio in the corrosion patina. No background subtraction was carried out because of the abrasive transference of the solid sample onto the graphite electrode, its surface becomes scratched and the ‘true’ background differs from the blank voltammogram recorded at unmodified electrodes. Prior modeling of copper/bronze coins electrochemistry assumed a variation of the tenorite/cuprite ratio with depth following a potential law. As a result, the ratio I(II)/I(I) varies on the value of I(I) as [8, 16, 48]:

Square wave voltammograms for sub-microsamples from the corrosion layers of (a) eurocent coin (three replicate experiments), (b–d) coins C1 (b), C3 (c) and C4 (d), attached to graphite electrode in contact with air-saturated 0.25 M HAc/NaAc aqueous buffer at pH 4.75. Potential scan initiated at 0.75 V vs. Ag/AgCl in the negative direction; potential step increment 4 mV; square wave amplitude 25 mV; frequency 5 Hz
A and α being two constants whose values are representative of the composition, manufacturing protocol, and corrosion level of the coin. Figure 3a depicts the plots of I(II)/I(I) vs. I(I) for the coins studied here based on data from five replicate voltammograms sampling in different regions of each coin. One can see that, in agreement with previous results [8, 16, 48], experimental data can be fitted to different tendency lines. The eurocent coins (squares) are confined to clearly lower I(II)/I(I) ratios, in agreement with their expected low proportion of tenorite. Coins C1 and C2 (solid squares) occupied an essentially common curve whereas coins C3–C6 (triangles) were distributed in a region having the higher I(II)/I(I) ratios. The corresponding double-logarithmic representation of I(II)/I(I) vs. I(I) for the archaeological coins is shown in Fig. 3b and permits their discrimination in close, but separate, tendency lines. One can see in this figure that the plots of log [I(II)/I(I)] vs. log [I(I)] show a satisfactory linearity permitting to define the ordinate at the origin (formally equal to log A) and the slope (given by the exponent α in Eq. (1)) as a parameters representative of each coin. Accordingly, these two parameters can be taken as representative of the current state of the metal patina, depending on the chemical composition of the original metal, the metallographic treatment used in the coin manufacturing, and the ‘corrosion history’ of the piece.

Representation of (a)i(II)/i(I) vs. i(I) and (b) log [i (II)/i(I)] vs. log [i(I)] for coins in this study. Data from five-seven replicate voltammograms such as in Fig. 2 sampling in different regions of each coin
EIS
Preliminary electrochemical experiments were performed using eurocent coins in contact with different electrolytes. As expected, EIS experiments in NaCl solutions using dissolved oxygen or Fe (CN)63−/4− as redox probes produced significant damage in the surface of the coins. In contrast, no significant (apparent) damage was observed using air-saturated NaClO4 aqueous solutions and mineral water at potentials between −0.2 and − 0.8 V vs. Ag/AgCl, which were used to test the possibility of using the reduction of dissolved oxygen as a redox probe. In these conditions, however, there is possibility of producing the reduction of copper compounds in the metal patina, thus altering the composition and structure of the same. As previously studied, such processes involve proton insertion associated to electron transfer, so that only occur at a significant rate at the time scale of voltammetric experiments in acidic electrolytes [8, 16, 49].
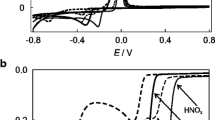
Although no visual appearance of surface damage was observed in EIS experiments at mineral water and NaClO4 aqueous solution, in order to discard the potential damage to the coin surface, cyclic voltammetric and controlled potential coulometric experiments were performed on eurocent coins at a time scale equivalent to that of EIS measurements. Pertinent data are shown in Fig. 4, where cyclic voltammograms and current/time curves recorded upon application of a constant potential of −0.65 V vs. Ag/AgCl for eurocent coins in contact with mineral water and 0.10 M NaClO4 are compared with those recorded in 0.25 M HAc/NaAc buffer at pH 4.75. In this electrolyte, the reduction of copper corrosion products (mainly cuprite) to copper metal followed by its subsequent stripping oxidation look likes intense, noisy responses. Such features result from the de-aggregation of the corrosion patina and possibly gas evolution, making this electrolyte unable to be used in EIS measurements, where stationary, equilibrium-like conditions are demanded.

a) Cyclic voltammograms (two potential cycles at potential scan rate 50 mV s−1) and (b) current/time curves recorded upon application of a constant potential of −0.65 V vs. Ag/AgCl for eurocent coins in contact with mineral water (red), 0.10 M NaClO4 (green) and 0.25 M HAc/NaAc buffer at pH 4.75 (black)
The charge passed in such experiments in both mineral water and 0.10 M NaClO4 at times below 1 min were 10–20 times lower than those passed in acetate buffer. Remarkably, no stripping peaks corresponding to the oxidation of copper metal formed in the reduction of copper corrosion products were detected in these electrolytes. This means that the unique Faradaic process occurring significantly at the time scale of EIS experiments in mineral water and 0.10 M NaClO4 is actually the reduction of dissolved oxygen (oxygen reduction reaction, ORR).
Figure 5 shows the Nyquist and Bode plots corresponding to the EIS measurements on a modern 5 eurocent coin in contact with air-saturated mineral water and 0.10 M NaClO4 at the OCP. The Nyquist representation consisted on a depressed capacitive loop. As expected, due to the different conductivity of mineral water and NaClO4 electrolytes, the absolute values of solution resistance, in principle given by the impedance at the extreme of high frequencies were clearly larger in mineral water than in 0.10 M NaClO4. The Bode plots of the (minus) phase angle vs. log (frequency) showed a maximum at intermediate frequencies while the representation of log (modulus of total impedance) vs. log (frequency) consisted of an s-shaped curve. These spectra can be satisfactorily modeled using a Randles-type circuit which is constituted by a solution resistance (Rs), a charge transfer resistance (Rct) in series with a Warburg element (W) and in parallel with a constant phase element (CPEdl) representative of the non-ideal behaviour of the double-layer capacitance (see Fig. S1a in Supplementary Information). This circuit has been used to study protective coatings on copper around OCP [50, 51]. Table S1 (Supplementary Information) summarizes the averaged values determined for such elements from simulations on three independent impedance spectra.

(a, b) Nyquist and (c–d) Bode plots from impedance measurements on eurocent coin in contact with (a, c) air-saturated mineral water and (b, d) air-saturated 0.10 M NaClO4 at the OCP. Black circles: experimental data points; Red circles: theoretical spectra modeled by the equivalent circuit in Fig. S1
Figure 6 compares the Nyquist and Bode plots corresponding to the EIS measurements on a modern five eurocent coins in contact with air-saturated mineral water and 0.10 M NaClO4 under the application of bias potentials of −0.4 V (black), −0.6 V (green), −0.8 V (red) vs. Ag/AgCl, all around the peak potential for the reduction of dissolved oxygen previously determined [8, 16]. The spectra exhibited relatively large dispersion at low frequencies, an effect which was particularly marked at −0.8 V vs. Ag/AgCl and again the impedances in mineral water were clearly larger than in 0.10 M NaClO4. Repeatability tests produced satisfactory results, with no significant changes in the EIS spectra for 3–5 replicate experiments on the same coin. The inclusion of a Warburg element in the equivalent circuits (see Fig. S1) is representative of the oxygen mass transfer through the electrolyte, and mainly, in the case of highly corroded coins, of the partially impeded oxygen transport through the microparticulate corrosion products to reach the metallic surface.

(a, b) Nyquist and (c–d) Bode plots from impedance measurements on eurocent coin in contact with (a, c) air-saturated mineral water and (b, d) air-saturated 0.10 M NaClO4 under the application of different bias potentials: −0.4 V (black), −0.6 V (green), −0.8 V (red) vs. Ag/AgCl
Such spectra were satisfactorily fitted to the equivalent circuit in Fig. S1b, used to describe cuprite [52] and self-assembled protective monolayers [53] on copper. These elements can be associated to the charge transfer through the metal patina displaying a resistive effect and a capacitive one in the patina/electrolyte interface. Fig. S3 in Supplementary information depicts the superimposed experimental and modeled Nyquist plots at a bias potential of −0.4 V in contact with mineral water and 0.10 M NaClO4.
Application of bias potentials between 0.2 and − 0.2 V produced impedance spectra relatively more complicated (see Fig. S2 in Supplementary Information) whose modeling can be satisfactorily made using the equivalent circuit in Fig. S1c, also used for describing metal corrosion a nonporous oxide layer or ‘barrier layer’ [54, 55].
To rationalize the recorded EIS, one can assume that, at bias potentials between −0.4 and − 0.8 V vs. Ag/AgCl, the impedance response occurred under approximately stationary conditions being dominated by the ‘forced’ charge transfer associated to the reduction of dissolved oxygen. This is a multi-step electron transfer process whose first stage in neutral and alkaline media is the one-electron reduction of O2 to anion radical superoxide which is followed by protonation and competing disproportionation/electron transfer processes [56, 57]. For our purposes, the relevant aspect to underline is that this process, in spite of this complexity, provides a net flux of charge transfer through the electrode/electrolyte interface. At bias potentials between 0.2 and − 0.4 V vs. Ag/AgCl, the ORR process does not occur significantly and charge separation/transfer is similar to that recorded under open circuit potential (OCP) conditions. In order to apply impedance measurements to archaeological metal objects, the above results suggested:
-
a)
Although both electrolytes provide comparable results in regard to repeatability and modeling of EIS measurements, it seems obvious that mineral water provides minor possibility of alteration so that it was used for studying archaeological artefacts.
-
b)
As far as the characterization of archaeological objects involves the determination of parameters dependent on the patina composition, structure and texture (roughness, porosity), EIS spectra at potentials between −0.4 and − 0.6 V vs. Ag/AgCl appeared as the most convenient for such purposes.
Figure 7 compares the Nyquist plots for the impedance measurements at −0.6 V vs. Ag/AgCl of coins a) C1, b) C6, c) C3 and d) C5 in contact with air-saturated mineral water. In the case of coins C1 and C2, both presenting a uniform patination, the representation corresponds to a depressed semicircle which is replaced, in the case of samples with gross uniform patination (C3 and C6), by two overlapping loops. Remarkably, the size of the loop at high frequencies increases on increasing the apparent degree of alteration of the coin, being higher for coins C4 and C5, displaying irregular gross alteration locally accompanied by pitting corrosion. Consistently, Bode plots (see Fig. S3 in Supplementary Information) displayed two maxima of the (minus) phase angle at frequencies between 500 and 100 Hz and between 5 and 10 Hz, respectively. These features indicated that there was (at least) two relaxation processes to be accounted in the impedance measurements. Although the dispersion of experimental points in the low frequency region was also enhanced on increasing the degree of alteration of the coin surface, thus making difficult the modeling, EIS measurements of coins C1 and C2 were satisfactorily fitted (see Fig. 7a) to the equivalent circuit represented in Fig. S1b, in agreement with previous data for eurocent coins. The impedance spectra of samples C3 and C6 (Fig. 7b, c) was fitted, however, to the equivalent depicted in Fig. S4a (Supplementary Information), containing a series combination of two parallel resistance plus capacitors (optionally replaced by a CPE) sets, inspired in more complicated circuits used to model the formation of oxide layers on copper electrodes [56, 58, 59].

Nyquist plots corresponding to impedance measurements of coins (a) C1, (b) C6, (c) C3 and (d) C5 in contact with air-saturated mineral water at a bias potential of −0.6 V vs. Ag/AgCl. Black circles: experimental data points; Red circles: theoretical spectra modeled by the equivalent circuits in Figs. S1b (a), S4a (b, c) and b (d)
Discussion
Figure 8 shows for a two-dimensional diagram containing the values of the ordinate at the origin and the slope of log [I(II)/I(I)] vs. log [I(I)] plots for a set of different eurocent coins with apparently equivalent smooth degree of alteration, and the archaeological coins studied here. One can see that the data points are approximately distributed along an inclined band, thus suggesting that both parameters are related. In this diagram, the coins C3 and C6, both displaying (see Fig. 1) high level of corrosion with local formation of agglomerates of green corrosion products, occupy a position in this band opposite to that of eurocents, whereas coins C1 and C2 are the nearest and coins C4, C5 fall in an intermediate region of the aforementioned inclined band.

Two-dimensional diagram containing the values of the ordinate at the origin (OO) and the slope (SL) of log [i(II)/i(I)] vs. log [i(I)] plots (see Fig. 3b) for coins in this study
In order to compare VIMP and EIS data, and due to the absence of a common equivalent circuit able to describe satisfactorily the different corrosion patterns, an empirical approach was used based on the use of φmax, logfmax and log|Zlow| as parameters representative of the impedance response of the corroded metal surfaces, where fmax represents the frequency at which the maximum (negative) phase angle, φmax, appears in the region of intermediate frequencies of the impedance spectra and |Zlow| the absolute value of the total impedance at the lowest tested frequency. This treatment was inspired by that described by Chen et al. [58] in their study of porous oxygen electrodes with discrete particles and was previously used for characterizing coin series with low degree of corrosion [8, 16, 48].
Figure 9 depicts the logfmax vs. log|Zlow| plots from data in replicate EIS experiments on the studied coins using air-saturated mineral water as the electrolyte and applying a bias potential of −0.6 V vs. Ag/AgCl. One can see in this figure that C1 and C2 defined a common tendency curve close, but separated from those of coins C4 and C5 and that of eurocent coins. In turn, these are clearly separated from the tendency curves of highly corroded coins C3 and C6 who also differed between them.

Variation of logfmax on log|Zlow| at different bias potentials for eurocent coins and nineteenth century coins in this study. From five independent replicate experiments in contact with air-saturated mineral water at a bias potential of −0.6 V vs. Ag/AgCl
These results can be considered as consistent with the idea that, when a similar corrosion pattern appears, EIS data can be used to discriminate between different monetary emissions. Thus, coins C4 (Spanish Republic, 1870) and C5 (Alphonse XII, Spain, 1877) displaying middle corrosion degree, produced impedance responses which can be separated using appropriate parameters. The same situation was obtained for highly corroded coins C3 (Spanish Republic, 1870) and C6 (Alphonse XII, Spain, 1877), as can be seen in Fig. 9.
It is pertinent to note that VIMP sampling was carried out on brown-black regions of the coins and is particularly responsive to the tenorite/cuprite composition of the secondary patina. In contrast, EIS experiments included also the regions presenting high local corrosion and were particularly representative, under our experimental conditions, not only of the composition of the corrosion layers (influencing the charge transfer impedance), but also on its homogeneity, thickness, porosity and roughness. Accordingly, there is possibility of discrepancies between both techniques in relation to their evaluation of the corrosion degree of the metallic surfaces.
Remarkably, the coins from different series appear to follow parallel, but different, ‘corrosion routes’ which can be seen in Fig. 10, in which the values of the (minus) maximum phase angle, φmax, recorded at intermediate frequencies are plotted vs. the logarithm of the frequency at which this maximum appears. Here, the eurocent coins fall in a region of the diagram separated from those occupied by aged coins which in turn become progressively separated from the eurocents as the degree of corrosion increases. Consistently with the foregoing considerations, the corrosion route defined for the sequence C1 → C5 → C3 coins (Alphonse XII 1877, grey arrow) differs from the corrosion route defined by the couple C4 → C6 of the Spanish Republic (1870, white arrow) coins (C4, C6), thus confirming the idea that the advance of the corrosion acts as an amplifier (in terms of electrochemical response) of the differences between different coinage series [8, 16].

Variation of the (minus) maximum phase angle recorded at intermediate frequencies on the logarithm of the frequency at which this maximum appears for impedance measurements at a bias potential of −0.6 V vs. Ag/AgCl for coins in this study. The ‘corrosion routes’ for Alphonse XII 1877 coins (C1, C3, C5) and Spanish Republic 1870 coins (C4, C6) are marked by grey and white arrows, respectively
All these results can be considered as indicative of the capability of obtaining satisfactorily stationary EIS responses of coins in non-aggressive mineral water and the possibility of using these measurements for assessing the degree and type of corrosion as well as for discriminating different monetary emissions under conditions of equivalent aging, confirming previous results using homogeneous series of coins [8, 16].
Conclusions
Voltammetry of microparticles measurements on sub-microsamples from the patina of eurocent coins and highly corroded nineteenth century copper/bronze coins in contact with aqueous acetate buffer-produced systematic variations of the signals for the reduction of tenorite and cuprite, grouping the coins for different corrosion patterns. Such variations were consistent with those established using electrochemical impedance measurements using the coins as a working electrode in contact with air-saturated mineral water. In this case, reasonably stationary responses were obtained mainly using the reduction of dissolved oxygen as a redox probe. The electrochemical techniques have proven to be a useful tool for the studies on ancient metals as they give information without compromising the conservation of the samples. In particular, the voltammetry gives information about the composition of the artefacts and its patina. Moreover, the voltammetric techniques can be more easily understood by no-specialist public in electrochemistry. On the other hand, EIS is a powerful and useful technique, especially regarding the corrosion issues. However, EIS can be of difficult interpretation for ancient and corroded metals. So, it is desirable a combination of both techniques as they are useful to give a comprehensive information about the artefact. In fact, data reported her indicate that it is possible to characterize different corrosion routes and discriminating, in favorable cases, different monetary productions, thus defining a potentially useful analytical tool for the study of highly altered metallic heritage.
References
Scott DA (1991) Metallography and microstructure of ancient and historic metals. Getty Conservation Institute in association with Archetype Books, Marina del Rey, p 155
Ingo GM, Balbi S, De Caro T et al (2006) Microchemical investigation of Greek and Roman silver and gold plated coins: coating techniques and corrosion mechanisms. Appl Phys A Mater Sci Process 83(4):623–629
FitzGerald KP, Nairn J, Skennerton G, Atrens A (2006) Atmospheric corrosion of copper and the colour, structure and composition of natural patinas on copper. Corros Sci 48(9):2480–2509
Robbiola L, Blengino JM, Fiaud C (1998) Morphology and mechanisms of formation of natural patinas on archaeological Cu-Sn alloys. Corros Sci 40(12):2083–2111
Chiavari C, Rahmouni K, Takenouti H, Joiret S, Vermaut P, Robbiola L (2007) Composition and electrochemical properties of natural patinas of outdoor bronze monuments. Electrochim Acta 52(27):7760–7769
Robbiola L, Hurtel L (1997) Standard nature of the passive layers of buried archaeological bronze - the example of two Roman half-length portraits. In: METAL 95: international conference on metals conservation. James and James science Publisher, Semuren-Auxois, pp 109–117
Serghini-Idrissi M, Bernard MC, Harrif FZ, Joiret S, Rahmouni K, Srhiri A, Takenouti H, Vivier V, Ziani M (2005) Electrochemical and spectroscopic characterizations of patinas formed on an archaeological bronze coin. Electrochim Acta 50(24):4699–4709
Di Turo F, Montoya N, Piquero-Cilla J et al (2017) Archaeometric analysis of Roman bronze coins from the Magna Mater temple using solid-state voltammetry and electrochemical impedance spectroscopy. Anal Chim Acta 955:36–47
Di Turo F, De Vito C, Coletti F et al (2017) A multi-analytical approach for the validation of a jellified electrolyte: application to the study of ancient bronze patina. Microchem J 134:154–163
Aucouturier M, Darque-Ceretti E (2007) The surface of cultural heritage artefacts: physicochemical investigations for their knowledge and their conservation. Chem Soc Rev 36(10):1605
Ingo GM, Riccucci C, Faraldi F, Pascucci M, Messina E, Fierro G, di Carlo G (2017) Roman sophisticated surface modification methods to manufacture silver counterfeited coins. Appl Surf Sci 421:109–119
Griesser M, Kockelmann W, Hradil K, Traum R (2016) New insights into the manufacturing technique and corrosion of high leaded antique bronze coins. Microchem J 126:181–193
Ingo GM, Balbi S, De Caro T et al (2006) Combined use of SEM-EDS, OM and XRD for the characterization of corrosion products grown on silver roman coins. Appl Phys A Mater Sci Process 83(4):493–497
Bernard MC, Joiret S (2009) Understanding corrosion of ancient metals for the conservation of cultural heritage. Electrochim Acta 54(22):5199–5205
Matthiesen H, Hilbert LR, Gregory D, Sørensen B (2004) Corrosion of archaeological iron artefacts compared to modern iron at the waterlogged site Nydam, Denmark. Eurocorr 2004:1–12
Doménech-Carbó A, Doménech-Carbó MT, Montagna E, Álvarez-Romero C, Lee Y (2017) Electrochemical discrimination of mints: the last Chinese emperors Kuang Hsü and Hsüan T’ung monetary unification. Talanta 169:50–56
Doménech-Carbó A, Doménech-Carbó MT, Capelo S, Pasíes T, Martínez-Lázaro I (2014) Dating archaeological copper/bronze artifacts by using the voltammetry of microparticles. Angew Chem Int Ed 53(35):9262–9266
Doménech-Carbo A (2017) Electrochemical dating: a review. J Solid State Electrochem 21(7):1987–1998
Doménech-Carbó A, Doménech-Carbó M, Martínez-Lázaro I (2008) Electrochemical identification of bronze corrosion products in archaeological artefacts. A case study. Microchim Acta 162(3-4):351–359
Di Turo F, Montoya N, Piquero-Cilla J et al (2018) Dating archaeological strata in the magna mater temple using solid-state voltammetric analysis of leaded bronze coins. Electroanalysis 30(2):361–370
Ramírez Barat B, Cano E (2018) In situ electrochemical impedance spectroscopy measurements and their interpretation for the diagnostic of metallic cultural heritage: a review. ChemElectroChem 5:2698–2716
Sharer Sahir Z, Sykes JM (2014) Effect of temperature on the impedance response of coated metals. Prog Org Coat 77(12):2039–2044
Amirudin A, DT AA, Thierry D (1995) Application of electrochemical impedance spectroscopy to study the degradation of polymer-coated metals. Prog Org Coat 26(1):1–28
Láng GG, Ujvári M, Bazsó F, Vesztergom S, Ujhelyi F (2012) In situ monitoring of the electrochemical degradation of polymer films on metals using the bending beam method and impedance spectroscopy. Electrochim Acta 73:59–69
Fatkullin AR, Parfenov EV, Yerokhin A, Lazarev DM, Matthews A (2015) Effect of positive and negative pulse voltages on surface properties and equivalent circuit of the plasma electrolytic oxidation process. Surf Coat Technol 284:427–437
Bonora PL, Deflorian F, Fedrizzi L (1996) Electrochemical impedance spectroscopy as a tool for investigating underpaint corrosion. Electrochim Acta 41(7-8):1073–1082
Mora N, Cano E, Polo JL, Puente JM, Bastidas JM (2004) Corrosion protection properties of cerium layers formed on tinplate. Corros Sci 46(3):563–578
Doménech-Carbó A, Doménech-Carbó MT, Peiró-Ronda MA, Martínez-Lázaro I, Barrio-Martín J (2012) Application of the voltammetry of microparticles for dating archaeological lead using polarization curves and electrochemical impedance spectroscopy. J Solid State Electrochem 16(7):2349–2356
Doménech-Carbò A, Capelo S, Piquero J et al (2016) Dating archaeological copper using electrochemical impedance spectroscopy. Comparison with voltammetry of microparticles dating. Mater Corros 67(2):120–129
Doménech-Carbò A, Domènech-Carbò MT, Martìnez-Làzaro I (2010) Layer-by-layer identification of copper alteration products in metallic works of art using the voltammetry of microparticles. Anal Chim Acta 680(1-2):1–9
Doménech-Carbó A, Lastras M, Rodríguez F, Cano E, Piquero-Cilla J, Osete-Cortina L (2014) Monitoring stabilizing procedures of archaeological iron using electrochemical impedance spectroscopy. J Solid State Electrochem 18(2):399–409
Dermaj A, Hajjaji N, Joiret S, Rahmouni K, Srhiri A, Takenouti H, Vivier V (2007) Electrochemical and spectroscopic evidences of corrosion inhibition of bronze by a triazole derivative. Electrochim Acta 52(14):4654–4662
Hassairi H, Bousselmi L, Khosrof S (2013) Evaluation of the inhibitive effect of benzotriazole on archeological bronze in acidic medium. Appl Phys A Mater Sci Process 113(4):923–931
Adriaens A, Dowsett M (2008) Time resolved spectroelectrochemistry studies for protection of heritage metals. Surf Eng 24(2):84–89
Di Turo F, Matricardi P, Di Meo C, Mazzei F, Favero G, Zane D (2018) PVA hydrogel as polymer electrolyte for electrochemical impedance analysis on archaeological metals. J Cult Herit. https://doi.org/10.1016/j.culher.2018.09.017
Di Fazio M, Di Turo F, Medeghini L et al (2019) New insights on medieval Provisini silver coins by a combination of non-destructive and micro-invasive techniques. Microchem J 144:309–318. https://doi.org/10.1016/j.microc.2018.09.016
Sluyters-Rehbach M, Sluyters JH (1979) On the meaning of the impedance concept in the case of an object that varies with time and in the case of a swept F frequency. J Electroanal Chem 102(3):415–419
Popkirov GS, Schindler RN (1993) Validation of experimental data in electrochemical impedance spectroscopy. Electrochim Acta 38(7):861–867
Macdonald DD, Sikora E, Engelhardt G (1998) Characterizing electrochemical systems in the frequency domain. Electrochim Acta 43(1-2):87–107
Bastidas JM, Polo JL, Torres CL, Cano E (2001) Study on the stability of AISI 316L stainless steel pitting corrosion through its transfer function. Corros Sci 43(2):269–281
Degrigny C, Guibert G, Ramseyer S, Rapp G, Tarchini A (2010) Use of E corr vs time plots for the qualitative analysis of metallic elements from scientific and technical objects: the SPAMT test project. J Solid State Electrochem 14(3):425–435
Vázquez-Huerta G, Ramos-Sánchez G, Rodríguez-Castellanos A, Meza-Calderón D, Antaño-López R, Solorza-Feria O (2010) Electrochemical analysis of the kinetics and mechanism of the oxygen reduction reaction on Au nanoparticles. J Electroanal Chem 645(1):35–40
Çirmi D, Aydin R, Köleli F (2015) The electrochemical reduction of nitrate ion on polypyrrole coated copper electrode. J Electroanal Chem 736:101–106
Scholz F, Meyer B (1996) Voltammetry of solid microparticles immobilized on electrode surfaces. Electroanal Chem 20:1–82
Scholz F, Schröder U, Gulaboski R, Doménech-Carbó A (2015) Electrochemistry of immobilized particles and droplets, 2nd edn. Springer International Publishing, Berlin-Heidelberg
Doménech-Carbó A, Labuda J, Scholz F (2013) Electroanalytical chemistry for the analysis of solids: characterization and classification (IUPAC technical report)*. Pure Appl Chem 85:609–631
Doménech-Carbó A, Doménech-Carbó MTM, Costa V (2009) Electrochemical methods in archaeometry, conservation and restoration, I. Springer Berlin Heidelberg, Berlin
Doménech-Carbó A, Doménech-Carbó MT, Álvarez-Romero C, Montoya N, Pasíes-Oviedo T, Buendía M (2017) Electrochemical characterization of coinage techniques the 17thCentury: the maravedís case. Electroanalysis 29(9):2008–2018
Redondo-Marugán J, Piquero-Cilla J, Doménech-Carbó MT, Ramírez-Barat B, Sekhaneh WA, Capelo S, Doménech-Carbó A (2017) Characterizing archaeological bronze corrosion products intersecting electrochemical impedance measurements with voltammetry of immobilized particles. Electrochim Acta 246:269–279
Rahal C, Masmoudi M, Abdelhedi R, Sabot R, Jeannin M, Bouaziz M, Refait P (2016) Olive leaf extract as natural corrosion inhibitor for pure copper in 0.5 M NaCl solution: a study by voltammetry around OCP. J Electroanal Chem 769:53–61
Grdeń M (2014) Interfacial capacitance of an oxidised copper electrode. J Electroanal Chem 713:47–57
Yi P, Dong C, Xiao K, Man C, Li X (2018) In-situ investigation of the semiconductive properties and protective role of Cu2O layer formed on copper in a borate buffer solution. J Electroanal Chem 809:52–58
Caprioli F, Martinelli A, Di Castro V, Decker F (2013) Effect of various terminal groups on long-term protective properties of aromatic SAMs on copper in acidic environment. J Electroanal Chem 693:86–94
Ribotta SB, La orgia LF, Gassa LM, Folquer ME (2008) Characterization of anodic films formed on copper in 0.1 M borax solution. J Electroanal Chem 624(1-2):262–268
Poljacek SM, Risovic D, Cigula T, Gojo M (2012) Application of electrochemical impedance spectroscopy in characterization of structural changes of printing plates. J Solid State Electrochem 16(3):1077–1089
Xu J, Huang W, McCreery R (1996) NewsReader project. J Electroanal Chem 410(2):235–242
Jang JH, Oh SM (2004) Complex capacitance analysis of porous carbon electrodes for electric double-layer capacitors. J Electrochem Soc 151(4):A571
Chen G, Waraksa C, Cho H et al (2003) EIS studies of porous oxygen electrodes with discrete particles-I. Impedance of oxide catalyst supports. J Electrochem Soc 150:E423–E428
Survila A, Surviliene A, Kanapeckaite S et al (2005) Oxide layers developed on copper electrodes in Cu (II) solutions containing ligands. J Electroanal Chem 582(1-2):221–229
Funding
Project CTQ2017-85317-C2-1-P, which is supported with Ministerio de Economía, Industria y Competitividad (MINECO) and Fondo Europeo de Desarrollo Regional (ERDF) funds, and Agencia Estatal de Investigación (AEI) and PhD grants of the Department of Earth Sciences, Sapienza University of Rome, are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 297 kb)
Rights and permissions
About this article

Cite this article
Di Turo, F., Parra, R., Piquero-Cilla, J. et al. Crossing VIMP and EIS for studying heterogeneous sets of copper/bronze coins. J Solid State Electrochem 23, 771–781 (2019). https://doi.org/10.1007/s10008-018-04182-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-018-04182-5