
Abstract
A study on the preparation and characterization of the potentiostatically prepared bismuth films (BiFs), in order to obtain satisfactory electroanalytical tool, is presented. BiFs formed on glassy carbon were characterized by electrochemical impedance spectroscopy (EIS) and scanning electron microscopy (SEM). The dependence of the BiFs properties upon electrolytes composition and electrochemical parameters are discussed and diagnostic criteria that allow estimation of the BiF morphology are proposed. Analytical performance data of the formed BiF electrodes were obtained by their application to the determination of glutathione (GSH) and folic acid (FA) using square-wave cathodic stripping voltammetry. The dependence of the analytical performance of the formed BiFs on their specific surface areas, along with their different morphology, is discussed. Adequate method and parameters for the electrochemical formation of optimal BiF, in order to fulfill the analytical requirements, are proposed. The best analytical performance was obtained with films formed from acetic buffer solution spiked with EDTA, as a consequence of the improved surface coverage and most arranged homogenous structure of the film. This electrode displays a linear response range toward GSH with estimated detection limit of 0.005 μM and sensitivity of 3.28 μA μM−1 for linear range of 0.01 to 0.1 μM. Also, the utilization of the BiF electrode for the determination of FA was demonstrated by direct electroreduction of FA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the last 15 years, bismuth has become an attractive and promising material for electroanalytical application due to its low toxicity and good operating cathodic potentials (thus excluding oxygen interference and hydrogen evolution over electroanalytical potential window). The electrodeposition of bismuth, on various substrates, has become the most commonly used electrode construction method, owing to the preparation protocol simplicity and electroanalytical performance of the electrodes prepared by this method (when compared to the mercury electrodes). However, the electroanalytical properties of bismuth film (BiF) are strongly influenced by its nature (structure, morphology, adherence, etc.) and this relationship has been described by several authors. A significantly improved bismuth film was formed from the acid solution of Bi(III), in the presence of bromide ions. By using this method, Krolicka et al. [1] formed an adhesive bismuth film on GC, characterized with large, lumpy, perpendicular crystallites, which showed good performance in the determination of Co2+. Moreover, in the presence of the same anion, Hutton et al. [2] prepared a uniformly and thoroughly coated bismuth film on a carbon fiber surface. Authors demonstrated a high long-term functional stability, as morphology and structure of the bismuth film remained unchanged even after 40 consecutive determinations of Co2+. Other additives were also used to obtain satisfactory Bi films. Therefore, Nunes et al. [3] performed the bismuth electrodeposition from hydrochloric acid, on a copper substrate, in the presence of citrate. The obtained film was composed of uniformly distributed aggregates of spherical particles (with a few nanometers in diameter) with a good adherence to the substrate and showed a good electroanalytical performance for lead determination. Using the same substrate, Ni et al. [4] performed the bismuth electrodeposition in the presence of various complexing ligands (tartaric acid, citric acid, and EDTA). They found that only the introduction of EDTA caused morphological changes of the BiF, from dendritic to flower-like structure (constructed by microrods). Hence, the dendritic-like structure was chosen for the electroanalytical determination of Cd2+.
In order to obtain a larger active area and lower background noise, some authors reported the construction and characterization of the porous film. This film can be formed by the electrodeposition of bismuth on a template which can be removed after the deposition, thus leaving the appropriate bismuth film. Such a bismuth film, deposited on a screen-printed carbon template containing CaCO3, was reported by Chen et al. [5]. This coated porous screen printed electrode exhibited an enhanced sensitivity for Pb2+ and Cd2+, due to less grainy and patchy individual metal crystallites. Also, Saturno et al. [6] prepared a micro/nanoparticle bismuth film electrode by bismuth electrodeposition on GC using an Al2O3 template. The improved electroanalytical performance for the determination of cadmium and chromium is supposed to derive from a dispersed bismuth film, characterized by a particulate appearance. Fibril-like bismuth nanostructures, on the porous Nafion polymer matrix, were prepared by Yang et al. [7]. The developed electrode was applied to determine Pb2+ and Cd2+.
Recently, the introduction and characterization of composite materials, in order to obtain a better analytical performance, have been reported. Bi-doped mesoporous carbon xerogel, confined in a chitosan matrix and deposited on a GC, was investigated by Fort et al. [8]. This type of electrode showed an excellent analytical behavior that allowed the determination of Cd2+ and Pb2+. Quyang et al. [9] obtained a Bi/single wall carbon nanotubes composite as the platform for ultratrace Cr6+ determination. Such a composite exhibited faster speeds of electron transfer, due to a uniform and smoother Bi film morphology.
As it can be seen, all the above mentioned reports relate to the determination of heavy metals. However, insight into the relationship between morphological characteristics of prepared Bi films and electroanalytical parameters in biomolecule determination is limited, due to poor reporting (only few articles are related to this issue). Cai et al. [10] used the principles reported by Krolicka [1] and deposited a bismuth film in the presence of the various halogen anions. Only the presence of Br− yielded a denser growth of smaller bismuth crystals, uniformly distributed on the GC surface. Authors used this film for the determination of Sudan I by square wave voltammetry. The same principle was adopted by Guzsvány et al. [11] for the determination of neonicotinoide insecticides. Although, after the deposition, only a quarter of the surface was covered with bismuth particles, by treatment of the deposited film in HCl and activation by potential cycling, authors generated densely and randomly arranged thin fibers around the nucleation centers. In addition, bismuth formed rhombohedral crystals resembling twigs, thus covering the entire substrate surface. Electrodeposited Bi-nanowires on GC were developed by Ananthi et al. [12] for the determination of FA by square wave voltammetry. Authors used hydrogen bubbles (electrodeposition was performed at −1.0 V in acetate buffer pH 4.5) as a “stagnant template” for obtaining the bundles of dendritic compact bismuth nanowires.
Considering the information presented above, it is difficult to comprehend the correlation between the film morphology and structure and a satisfactory analytical performance. As it can be seen, excellent analytical properties (even for identical analytes) were obtained with a different morphology and structure of bismuth films. This issue seems to be more specific and depends on the nature of analyte, its pH, analytical procedure, electrode substrate, electrode construction, possible collateral reaction, etc. However, some general conclusions can be derived: (a) electrodepositions performed in the presence of complexing additive resulted in a more homogeneous and uniformly distributed films; (b) such films showed better adhesion to the substrate surface; (c) it is desirable to obtain a highly developed bismuth surface in order to increase the analyte amount that can be accumulated (as most methods are stripping-based ones).
Although bismuth film electrodes are used in electrochemical determination of some pharmaceuticals (daunomycine [13], aminosalycilate [14], glucose [15, 16], sulfathiazine [17], vitamin B12 [18], sildenafil citrate-Viagra [19], and N-acetilcysteine [20]), insecticides and herbicides (paraquat [21], neonicotinoid insecticide [11], and atrazine [22]) and aromatic nitro-derivatives (2-nitrophenol [23], azo dye Sunset Yellow [24], and Sudan I [10]), their relevance for folic acid determination has been very poorly exploited. In addition, the research of sulfur containing organic molecules with electrodes based on bismuth or bismuth compounds was reported only by a few contributors [20, 25–27].
Folic acid (FA) is an essential water-soluble vitamin, very important for DNA and RNA synthesis and cell growth (important during rapid fetal growth in pregnancy), proper functioning of the brain, mental and emotional health, the conversion of some amino acids, and controlling homocysteine concentration in blood (which is associated with heart diseases). Together with the vitamin B12, FA is required for creating new blood cells and its deficiency can lead to anemia [28].
Glutathione (GSH) is a biologically active thiol, which plays an important role in the scavenge of physiological free radicals. Additionally, changes in intercellular GSH concentrations have been correlated with patients suffering from Alzheimer’s disease, Parkinson’s disease, diabetes mellitus, atherosclerosis, arthritis, epilepsy, as well as numerous cancer types [29].
FA and GSH are very reactive in the oceanic water column, as both have a role in bioactivity [30]. Also, FA has a well-known role in homocysteine metabolism, resulting in GSH as final product [31]. As it can be seen, it is very important to develop an analytical method for the determination of these species, separately [30] or simultaneously [31].
In this paper, we report the preparation of the bismuth film electrodes on a glassy carbon substrate under different conditions. Preliminary investigation on the relevance of such prepared films for determination of GSH and FA is presented. Films were prepared at a different pH, with or without the presence of a complexing agent (EDTA). Furthermore, the influence of other parameters on the films is discussed, in the light of a possible analytical application. These parameters were evaluated using the electrochemical impedance spectroscopy (EIS) and scanning electron microscopy (SEM). The obtained results revealed a strong dependence of BiF electrode analytical performance on its specific surface area and morphology.
Experimental
Chemicals, solutions, and apparatus
All solutions were prepared from analytical grade chemicals. Solutions of nitric acid, sodium acetate, acetic acid, and EDTA, all purchased from Kemika (Zagreb, Croatia), were prepared by dissolution in double distilled water. Stock solution of the bismuth nitrate (0.02 M) was prepared by dissolution of 99.99 % Bi(NO3)3 × 5H2O (Sigma Aldrich, St. Louis, MO, USA) in 0.5 M nitric acid. Solution of folic acid (FA) and glutathione (GSH) (both from Merck, Darmstadt, Germany) was prepared daily, by dissolution of appropriate amount of the FA or GSH in double distilled water, previously deaerated with N2. All experiments were carried out at 25 °C, controlled by thermostat (Huber CC1, Offenburg, Germany).
An electrochemical cell with saturated calomel electrode (SCE) as reference, Pt plate as auxiliary and glassy carbon electrode (GCE) (2 mm in diameter) (Metrohm, Herisau, Switzerland) as working electrode, were used. The bismuth bulk electrode (5 mm in diameter) (BiE) was made from a polycrystalline bismuth rod (Goodfellow Cambridge Ltd., UK), encapsulated in a laboratory built Teflon housing, that was equipped with a stainless steel piston to make the electrical contact.
Before use, GCE, which served as substrate electrodes in all experiments, were mechanically polished with emery paper (2000 grit) followed by polishing with alumina powder down to 0.05 μm. After polishing, electrodes were treated ultrasonically in redistilled water, rinsed with ethanol, and finally rinsed with redistilled water. In order to achieve a reproducible and active electrode surface, the electrodes were repetitively cyclized (50 cycles) in 0.5 M HNO3, over potential range −1.0 V to 1.0 V (scan rate of 100 mV s−1). Prior cyclization electrodes were first kept at −1.2 V for 60 s, than at +1.5 V for 60 s.
Electrochemical behaviors and potentiostatic electrodepositions of bismuth were performed in different electrolytes (0.5 M HNO3 or 0.1 M acetate buffer (pH 4.5)) at Bi(III) concentration of 1 mM. When experiments were performed in the presence of EDTA, electrolytes were spiked with 1 mM EDTA and solutions were stirred and allowed to quiescent, after measurements were performed. All electrochemical measurements were carried out with potentiostat (Autolab PGSTAT 302N), connected to PC and driven by GPES 4.9 Software (Eco Chemie).
Impedance spectra were recorded in 0.5 M nitric acid, using a Solartron SI 1287 electrochemical interface and a Solartron SI 1255 frequency response analyzer connected to a PC. Impedance spectra were recorded at the potential of the evolution of hydrogen in nitric acid (E = −0.9 V), within the frequency range of 100 kHz–30 mHz with an ac signal ±10 mV. Impedance data were fitted by a suitable electrical equivalent circuit (EEC) model, employing the complex non-linear least squares (CNLS) fit analysis offered by the Solartron ZView® software. The fitting quality was evaluated by the chi-squared and relative error values, round 10−4 and below 5%, respectively, indicating that the between the proposed EEC model and the experimental, data were in good agreement.
For microscopic study, after electrodeposition, BiF electrodes were rinsed carefully in redistilled water, shortly dried in a nitrogen atmosphere then transferred to a microscope chamber. The surface morphology of the BiF electrodes was studied on Vega II LSH (TS 5130 LS) scanning electron microscope (Japan Electron Optics Laboratory, Japan). Obtained images were quantified using ImageJ Program (Rasband, U.S. NIH, Bethesda, Maryland).
For electroanalytical determination of the GSH and FA, ex situ BiF electrodes, prepared in acetic buffer solutions under various conditions, were used. Accumulation and the stripping measurements were carried out in deoxygenated solutions, under pure nitrogen atmosphere, and the parameters were determined by optimization. The determination of GSH was done as follows: prepared BiF electrode was washed and transferred rapidly to the acetate buffer solution. After addition of appropriate amount of GSH into 0.1 M acetate buffer solution (pH 4.5), the accumulation has been performed at cathodic potential (−0.35 V) for 60 s in stirred solution, followed by a 60-s accumulation in quiescent solution. A square wave cathodic stripping voltammetry (SWCSV) was performed in quiescent solutions from −0.35 to −0.8 V, with potential scan frequency (f) of the 10 Hz, pulse height (ΔE p) of the 100 mV, and potential increment (ΔE s) of the 20 mV.
The determination of FA was done as follows: prepared BiF electrode was washed and transferred rapidly to the acetate buffer solution. After addition of appropriate amount of FA into 0.1 M acetate buffer solution (pH 4.5), the accumulation (physical adsorption) has been performed at cathodic potential (−0.4 V) during 120 s in stirred solution, followed by an accumulation for 60 s in quiescent solution. A square wave cathodic stripping voltammetry (SWCSV) was performed in quiescent solutions from −0.35 to −0.8 V, with potential scan frequency (f) of the 30 Hz, pulse height (ΔE p) of the 100 mV and potential increment (ΔE s) of the 20 mV.
Before determination, a background response was recorded at same condition as mentioned above.
Results and discussion
Cyclic voltammetry
The electrochemical behaviors of GCE, in different electrolytes containing Bi(III), were investigated and corresponding cyclic voltammograms are presented in Fig. 1. The voltammogram obtained in nitric acid show well-defined cathodic and anodic peaks, which correspond to the bismuth deposition and dissolution, respectively. The voltammogram obtained in the acetate solution (pH 4.5) reveal a slight shift of the cathodic peak current potential toward negative values. This behavior is expected since at this pH, the main existing modality of the dissolved Bi(III) is BiO+. The addition of EDTA, in both systems, produces another cathodic peak, which indicates that the extraction of Bi3+ is now controlled by two different processes. It is reported that EDTA forms a stabile complexes with Bi(III), over a wide pH range, with different complexes form [32] which are presented in Eqs. (1) and (2):

a Cyclic voltammograms obtained at GCE in different electrolytes. b Enlargement of cathodic branch of cyclic voltammograms presented in Fig 1a. All solutions contained 1 mM Bi(III). Scan rate 25 mV s−1
However, since at pH 4.5 BiO+ is the main form of dissolved Bi(III), reaction (3) must be considered.
By the combination of reactions (2) and (3), complex formation reaction at pH 4.5 has a form:
As the cathodic current is controlled by free Bi3+, the first cathodic peak (A), in the resulted voltammogram obtained in HNO3 spiked with EDTA, can be attributed to the fast reduction (controlled by kinetics of the reaction (1)) of free Bi3+ near the electrode surface. As this peak is not clearly defined, indicating an important role of the complex diffusion toward electrode surface or the complex decomposition kinetics. As a result, two consecutive cathodic peaks can be noticed: (A) and (B).
In contrast, the voltammogram obtained in acetate buffer containing EDTA show two clearly defined reduction peaks. The first cathodic peak (A`) can be explained in the same manner as in the case of peak (A). However, there is a discrepancy in complex formation constant of reaction (4) and relatively high reduction signal of A`. Although electrodeposition kinetic of the Bi(III) is out of scope of this article, relatively high BiO+ concentration could be attributed to the slow kinetic of the formation of [Bi(EDTA)]−, as its formation is preceding by decomposition of the BiO+ which is less favored at increased pH values. The peak (B`) at −0.67 V can be attributed to the bismuth deposition, controlled by equations (4) and (3). Thus, a large potential difference between peaks (B`) and (B) is not surprising, as the extraction of Bi3+, in the presence of EDTA, follow more complex mechanism (together with a larger formation constant) than its extraction from HNO3, where deposition is controlled only by Eq. (1).
However, in both cases, a decreasing in the cathodic current, in the presence of EDTA, suggests the formation a bit more organized Bi nanostructure on GCE.
Scanning electronic microscopy
Insight into the morphology and the structure of electrodeposited particles were obtained by SEM. Based on the information obtained from CV, the bismuth films in HNO3 were prepared at −0.2 V and more negative potentials, during 60 s. The recorded micrographs are represented in Fig. 2. The electrodeposition performed at −0.2 V has suffered from insufficient electrode coverage—only 10 %, so this micrograph is not shown. As it can be seen, a potential shift toward more negative values leads to the formation of the tiny crystals (inset (a) of Fig. 2), which have a tendency to agglomerate at potential −0.7 V (inset (b) of Fig. 2). As it is presented in Table 1, at applied deposition potentials, partial GC surface coverage was achieved. Also, it must be pointed out that the film prepared at −0.9 V was non-adhesive and mechanically unstable. This behavior is not surprising, as at this potential, the electrodeposition of bismuth is accompanied by hydrogen evolution [33]. In order to increase surface coverage, the electrodepositions at −0.7 V, at extend deposition times (300 and 600 s), were performed. The micrograph of the film, prepared at deposition time for 600 s, is presented in Fig. 2. This film consisted of a large number of crystals of porous structure and poor stability.

SEM image of the bismuth films formed in 0.5 M nitric acid containing 1 mM Bi(III) at −0.7 V for 600 s. Inset: (a) at −0.4 V for 60 s; (b) at −0.7 V for 60 s
In addition, SEMs of the films formed in acetate buffer are presented in Fig. 3. Unlike porous deposit structure, obtained when electrodeposition was performed in HNO3, the film formed in acetate buffer at −0.7 V was characterized by irregular dendritic crystals (around 1 μm), resulting in a thick, quite uniform, non-porous layer (inset (a) of Fig. 3). The prolongation of deposition time up to 600 s (at −0.7 V) leads to almost totally covered GC surface (see Table 1). Contrary, at the same conditions and at −0.4 V, the GC surface was less covered, while the obtained film at −0.9 V suffered from spongy like morphology (inset (b) of Fig. 3).

SEM image of the bismuth films formed in 0.1 M acetate buffer solution (pH 4.5) containing 1 mM Bi(III) at −0.7 V for 600 s. Inset: (a) at −0.7 V for 60 s; (b) at −0.9 V for 60 s
Finally, the micrographs obtained from films formed in acetate buffer, spiked with EDTA, are presented in Fig. 4. As it can be seen, bismuth is in crystalline form, characterized with a large flakes-like dendritic structure. Also, an increase in surface coverage was obtained at −0.9 V, especially after prolonged deposition time (see Table 1).

SEM image of the bismuth films formed in 0.1 M acetate buffer solution (pH 4.5) containing 1 mM Bi(III) spiked with 1mM EDTA at −0.9 V for 600 s. Inset: (a) at −0.7 V for 60 s: (b) at −0.9 V for 60 s
As it mentioned, poor stability and porous structure of the bismuth deposition was attributed to the hydrogen evolution. Almost identical behaviors, as in the case when electrodepositions were performed in nitric acid, can be expected for bismuth films prepared in nitric acid spiked with EDTA. Indeed, as these films were formed at even more negative potentials (from −0.4 V), there were inevitably influenced by hydrogen evolution producing even poorer bismuth films (particularly in insufficient surface coverage), thus micrographs of these films were not recorded.
Also, a ratio of the total electrodeposition charge and the extent of the surface coverage can be used to the estimation of the crystal growth phenomenon. As it can be seen from the values presented in Table 1, it seems that the vertical growth of crystals is favored upon electrodepositions were performed in HNO3, while in acetate buffer, the lateral growth dominates.
As it is shown, SEM analyses revealed very high dependence of the bismuth film morphology upon electrolytes composition and parameters of the electrodeposition. The significant surface morphology improvements, together with increment of the surface coverage, were obtained when acetate buffer solution was spiked with EDTA.
Electrochemical impedance spectroscopy
It is well known that electrochemical impedance spectroscopy (EIS) can provide the information about surface coverage. Thus, EIS, coupled with a real time hydrogen evolution, was performed in order to gain further insight into the extent of surface coverage with bismuth. Based on the results of a preliminary study, evolution of the hydrogen was performed at the −0.9 V. For gathering reliable information, impedance spectra of a four different electrodes were recorded: the uncovered glassy carbon (GC), two BiF electrodes prepared in different electrolytes and the bismuth bulk electrode (BiE). The obtained results are presented in Fig. 5. The impedance spectra obtained for the GC and BiE can be considered as two ultimate cases: a bare electrode and a surface of GC completely covered with bismuth, respectively. The impedance spectra were evaluated using two different equivalent electrical circuits (EEC) presented in Fig. 5c and the results of the fitting procedure are given in Table 2. The first time constant arising from the charge transfer resistance (R ct) (due to the hydrogen evolution) in parallel with the double layer capacitance is represented by the parameters n and Q dl, while the second time constant can be described by the surface-related kinetic parameters concerning an evolution of hydrogen on the GC, as it probably follows Volmer-Heyrovsky mechanism [34]. These parameters are represented as pseudoresistance (R p) and pseudocapacitance represented by the parameters n and Q p. The obtained results indicate the possibility of the two different mechanisms of the hydrogen evolution on two different substrates. In addition, the spectra recorded for the BiF electrodes (prepared at a deposition time of 60 s) represent intermediate cases, where the surfaces are not completely covered with bismuth. This phenomenon is more distinguished in the Bode plots (see phase angles in Fig. 5b). As it can be seen, from the results obtained by fitting (Table 2), hydrogen evolution at BiF electrodes takes place on both substrate (deposited Bi and bare GC), simultaneously. This indicates insufficient coverage of the BiF electrodes. The fitted values (Table 2) reveal no difference in the charge transfer resistance, but in the pseudoresistance and the pseudocapacitance. These results suggest the kinetic limitation of hydrogen evolution on the bare GCE but not on the bismuth electrode (lack of the second time constant) and can be used as criterion of the surface coverage extent. Furthermore, as evolution of hydrogen is feasible at the bismuth surface, a porous structure of bismuth film prepared in HNO3, at −0.7 V, is not surprising.

Impedance spectra (Nyquist (a) and Bode (b) plots), recorded in 0.5 M nitric acid at −0.9 V, for different electrodes: GCE (squares); BiF electrode formed in nitric acid at −0.7 V for 60 s (circles); BiF electrode formed in acetate buffer at −0.7 V for 60 s (triangles up); BiE (triangles down). Nyquist plot for BiE is presented in the inset of Fig. 5a. Unfilled symbols represent values of phase angle vs. frequency. Equivalent electrical circuit models used for analysis (c)
Interestingly, R p values (Table 2), obtained for the BiF electrode formed in nitric acid and for the BiF electrode formed in acetate buffer, seem to be in discrepancy with values of the surface coverage obtained by SEM. However, based on the information about the morphology of the prepared films, this can be explained by a larger specific surface area in the case of the BiF obtained from HNO3. This indicates growth of the tip of the bismuth lump rather than the sides (lateral) spreading, resulting in the larger specific area but not in the surface coverage. Furthermore, Bode plot for the BiF electrodes, formed in acetate buffer at different deposition times, are presented in Fig. 6. The obtained data were fitted with the EEC presented in Fig. 5c, and the results are given in Table 3. The comparison of the kinetic parameter values suggests that a completely covered surface can be obtained at prolonged deposition times, as the second time constant disappears. This is in accordance with the surface coverage values obtained with SEM (Table 1). In addition, a similar spectra and behavior were observed for the deposits prepared in acetate buffer (at −0.9 V) in the presence of EDTA (see Fig. 6 and fitted data in Table 3).

The Bode plots, recorded in 0.5 M nitric acid at −0.9 V for BiF electrodes, formed in acetate buffer at −0.7 V, at different deposition times. The plot obtained for BiF electrode formed in acetate buffer spiked with 1 mM EDTA at −0.9 V for 600 s is represented with diamonds symbol. Unfilled symbols represent values of phase angle vs. frequency. Equivalent electrical circuit models used for analysis in Fig. 5c
Analytical applicability
Based on the above discussion and previous reports [4, 35], in order to compare its analytical potential, two films were chosen: one prepared from acetate buffer and the other formed in acetate buffer spiked with EDTA, both deposited under the optimized condition (t = 600 s and E = −0.7 or −0.9 V). In the present work, we employed SWCSV method to detect GSH. Determinations were performed in 0.1 M acetate buffer (pH 4.5) Before SWCSV measurements, the working potential window was determined by cyclic voltammetry (not shown), in the solution containing GSH. According to an analysis of obtained data, for the accumulation potential, E acc = −0.35 V was chosen. At this potential, the formation of Bi(GS)3 is expected to occur at the film surface, according equation (5).
The SWCS voltammograms were obtained by the standard addition method, and curves are background-subtracted currents. The calibration plots of the BiF electrodes toward GSH were derived from the obtained voltammograms, and the results are shown in Fig. 7. Other key operational parameters that influence on the peak current (I p) were established by optimization and their values are given in the “Experimental” section. The plots show that among all prepared films, the BiF prepared at −0.9 V, in the presence of EDTA, is highly sensitive for GSH—2.97 μA μM−1. The plot of the reduction peak current vs. GSH concentration is linear over the concentration range 0.1–1 μM of GSH. Furthermore, the linear range can be extended toward lower concentrations (Fig. 8), resulting with an increase in the analytical sensitivity (3.28 μA μM−1). The calculated detection limit, based on 3σ criterion [36], was 0.005 μM. The somewhat lower sensitivity for the higher concentration range (0.1–1 μM), with linearity preserving, can be attributed to the increased amount of Bi(GS)3(ads), accumulated at electrode surface, resulting in its saturation, as it was reported for the determination of cysteine on random array carbon fiber microelectrodes [20].

Voltammograms (without background subtraction) obtained for the GSH on BiF electrodes, prepared in 0.1 M acetic buffer, at different conditions: (A and circles) at −0.7 V; (B and squares) at −0.7 V in the presence of 1 mM EDTA; (C and triangles up) at −0.9 V; (D and triangle downs) at −0.9 V in the presence of 1 mM EDTA. In the middle: corresponding calibration plots (with background subtraction)

The SWCSVs obtained for the GSH, together with background response on BiF electrode prepared at −0.9 V in the presence of EDTA. The inset shows corresponding calibration plot. Experimental conditions are given in “Experimental” section
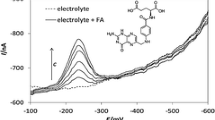
In addition, the utilization of the BiF electrode for the determination of FA was demonstrated by electroreduction of FA on the prepared BiF. In order to increase the sensitivity, prior to the FA electroreductions, adsorptive preconcentrations were performed. According to the preliminary results (not shown) for adsorption potential, E ads = −0.4 V was chosen. The mechanisms of the adsorptive preconcentration and electroreduction of FA have been explained by Le Gall and van der Berg [37] and Ananthi et al. [12]. The SWCS voltammograms for various concentrations of FA, together with the corresponding calibration plots, are presented in Fig. 9. The change in slope sensitivity (with linearity preserving) can be attributed to the identical process as in the case of determination of the GSH. However, the FA reduction potential (∼−0.6 V) is close to the potential used for determining of GSH, thus excluding the possibility of the simultaneously determination of these two species.

The SWCSVs obtained for the FA, together with background response on BiF electrode prepared at −0.9 V in the presence of EDTA. The insets show corresponding calibration plots for two different concentration ranges. Experimental conditions are given in “Experimental” section
The intra-day repeatability of the proposed method using BiF electrode was examined by ten successive measurements of GSH in solution containing 2 μM analyte at the same film. Obtained relative standard deviation, RSD = 11.0 %, indicates good repeatability. The reproducibility (4.9%) was evaluated using five freshly prepared bismuth film over 5 days. Under optimized experimental conditions, the reproducibility of the proposed methods for the determination of FA at the same ex situ formed BiF in regular intervals within 3 h was examined. The limiting factor for the determination of FA is possible degradation after a few hours of standing on light in acidic solution, which rapidly increases after 3 h. Measuring signal response after ten repeated measurements in a solution containing 2 μM was obtained relative standard deviation (RSD) of 1.4 %, which indicates good repeatability and stable response and opens the possibility of using BIF electrode for the electroanalytical purpose.
Since this work presents preliminary results, above-mentioned issues, together with other parameters of analytical performance, are out of scope of this study. Research related to alternative electrolytes composition and analytical protocol, which might address these issues more effectively, in order to attain a practical applicability of the BiF electrode, like determination of GSH and FA in real samples, are in full progress.
Conclusion
-
1.
The results presented in this study reveal significant dependence of the bismuth film morphology upon electrolyte composition and the parameters of electrodeposition. Generally, when electrodeposition had been performed in acetic buffer (pH 4.5), thick, uniform, non-porous layer was obtained. An addition of the complexing ligand (EDTA) into the acetic buffer yielded flakes-like dendritic structure. Generally, the films prepared in HNO3 suffered from porous structure and poor stability.
-
2.
Information obtained from impedance analysis suggests improved surface coverage in the case of the films prepared in acetic buffer, when compared with films obtained from HNO3. This data are in accordance with SEM images. In addition, data obtained from SEM and EIS suggest domination of lateral growth for the films obtained in acetic buffer, while the films prepared in HNO3 were characterized with vertical growth. Therefore, electrodepositions performed in acetic buffer significantly increase the surface coverage.
-
3.
The preliminary study on the relevance of BiF electrode for the determination of GSH revealed that film prepared in acetate buffer spiked with EDTA exhibits attractive stripping performance and provides enhanced sensitivity. In addition, utilization of BiF electrode in determination of FA is demonstrated.
-
4.
As one of the major objectives of the present study was to determine relationship between morphological characteristics of the film and its analytical performance, from obtained results, it can be concluded that improved electroanalytical performance of the BiF is supposed to benefit from the higher extent of the surface coverage, characterized with crystalline dendritic structure of the bismuth.
References
Krolicka A, Bobrowski A (2004) Electrochem Commun 6:99–104
Hutton EA, Hocevar SB, Ogorevc B (2005) Anal Chem Acta 537:285–292
Nunes LMS, Faria RC (2008) Electroanal 22:2259–2263
Ni Y, Zhang Y, Zhang L, Hong J (2011) Cryst Eng Comm 13:794–799
Chen C, Niu X, Chai Y, Zhao H (2009) Sens Actuat B Chem 178:261–269
Saturno J, Valera D, Carrero H, Fernandez L (2011) Sens Actuat B Chem 159:92–96
Yang D, Wang L, Chen ZL, Megharaj M, Naidu R (2014) Electroanalysis 26:639–647
Fort CI, Cotet LC, Vulpoi A, Turdean GL, Danciu V, Baia L, Popescu IC (2015) Sens Actuat B Chem 220:712–719
Ouyang RZ, Zhang WY, Zhou SL, Xue ZL, Xu LN, Gu YY, Miao YQ (2013) Electrochim Acta 113:686–693
Cai J, Zhou X, Tu Y, Feng G, Huang C (2012) Adv Mat Lett 3:87–91
Guzsvány V, Papp Z, Zbiljic J, Vajdle O, Rodic M (2011) Molecules 16:4451–4466
Ananthi A, Kumar SS, Phani KL (2015) Electrochim Acta 151:584–590
Duwensee H, Adamovski M, Flechsig GU (2007) Int J Electrochem Sci 2:498–507
Nigovic B, Simunic B, Hocevar S (2009) Electrochim Acta 54:5678–5683
Anık U, Timur S, Cubukcu M, Merkoci A (2008) Microchim Acta 160:269–273
Daniele S, Battistel D, Bergamin S, Bragato C (2010) Electroanalysis 22:1511–1518
Campestrini I, de Braga OC, Vieira IC, Spinelli A (2010) Electrochim Acta 55:4970–4975
Kreft GL, de Braga OC, Spinelli A (2012) Electrochim Acta 83:125–132
Sopha H, Hocevar SB, Pihlar B, Ogorevc B (2012) Electrochim Acta 60:274–277
Brinić S, Vladislavić N, Buzuk M, Bralić M, Solić M (2013) J Electroanal Chem 705:86–90
Figueiredo-Filho LCS, dos Santos VB, Campos Janegitz B, Guerreiro TB, Fatibello-Filho O, Faria RC, Marcolino-Junior LH (2010) Electroanalysis 22:1260–1266
Figueiredo-Filho LCS, Azzi DC, Janegitz BC, Fatibello-Filho O (2012) Electroanalysis 24:303–308
Hutton EA, Ogorevc B, Smyth MR (2004) Electroanalysis 16:1616–1621
Krolicka A, Bobrowski A, Zarebski J, Tesarowicz I (2014) Electroanalysis 26:756–765
Baldrianova L, Agrafiotou P, Svancara I, Vytras K, Sotiropoulos S (2008) Electrochem Commun 10:918–921
Nosal-Wiercinska A (2012) J Electroanal Chem 681:103–108
Nosal-Wiercinska A (2011) J Electroanal Chem 662:298–305
Yakubu S, Muazu J (2010) Der Pharmacia Sinica 1:55–58
Harfield J, Batchelor-McAuley C, Compton RG (2012) Analyst 137:2285–2296
Le Gall AC, Van Den Berg CMG (1998) Deep-Sea Research I 45:1903–1918
Raoof JB, Teymoori N, Khalilzadeh MA, Ojani R (2015) Mater Sci Eng C Mater Biol Appl 47:77–84
Stavila V, Davidovich RL, Gulea A, Whitmire KH (2006) Coord Chem Rev 250:2782–2810
Yang M (2011) J Mater Chem 21:3119–3124
Newman J, Thomas-Alyea KE (2004) Electrochemical systems. Wiley, New Jersey
Jiang S, Huang YH, Luo F, Du N, Yan C-H (2003) Inorg Chem Commun 6:781–785
Mocak J, Bond AM, Mitchell S, Scollary G (1997) Pure Appl Chem 69:297–328
Le Gall AC, Van Der Berg CMG (1993) Anal Chim Acta 2872:459–470
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vladislavić, N., Buzuk, M., Brinić, S. et al. Morphological characterization of ex situ prepared bismuth film electrodes and their application in electroanalytical determination of the biomolecules. J Solid State Electrochem 20, 2241–2250 (2016). https://doi.org/10.1007/s10008-016-3234-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3234-3