
Abstract
This work is a study of 5-amino-3-nitro-1,2,4-triazole (ANTA), 3-nitro-1,2,4-triazol-5-one (NTO), and nitrated derivatives of ANTA and NTO. RDX and TNT were studied for comparison. ANTA and NTO are low-sensitive high explosives with detonation properties comparable to 2,4,6-trinitrotoluene (TNT) and 1,3,5-trinitroperhydro-1,3,5-triazine (RDX). We showed previously that nitrated NTO and ANTA compounds, when used in a glycidyl azide polymer (GAP) matrix in rocket propellants, could give impulses above 2600 m/s and that the oxygen balance is positive. If used in aluminized explosives, the heat of detonation may be increased to a practical level significantly above RDX/aluminum compositions. Here, we use two different methods for sensitivity and two density functional theory functionals, B3LYP and M06-2X with the 6-31G(d) basis set, together with the complete basis set method CBS-4M. Calculations indicate that most of the nitrated derivatives have nearly equal sensitivity to RDX. Significantly different bond dissociation energies in the nitrimino functional group are predicted, although most models give much the same result.
Similar content being viewed by others
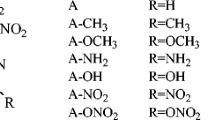

Avoid common mistakes on your manuscript.
Introduction
The high energetic triazoles 5-amino-3-nitro-1,2,4-triazole (ANTA) and 3-nitro-1,2,4-triazol-5-one (NTO) have high densities, and their detonation pressure and velocity are comparable to those of 1,3,5-trinitroperhydro-1,3,5-triazine (RDX). Nevertheless, ANTA and NTO have low sensitivity and might therefore be attractive materials in compositions where this property is important. Nitrated ANTA and NTO compounds might be even more interesting in this respect, due to the improved oxygen balance and enthalpy of formation of such derivatives. As long as the sensitivity and the thermal stability are tolerable, they may be suitable as fillers/oxidizers in minimum-smoke rocket propellant and in aluminized explosive compositions.
One of the most frequently used sensitivity measures is that of impact sensitivity. The test is performed by dropping a mass, most frequently 2.5 kg, on a sample, and determining the height from which 50% of the drops lead to reaction. The sensitivity is reported either as the drop height in centimeters or as the corresponding impact energy in Joules. Some parameters assumed to have an effect on the sensitivity have indeed been calculated, e.g., Mulliken charges, electrostatic potentials, and bond dissociation energies [1,2,3,4,5,6,7], but a general theory for impact sensitivity has not yet been introduced. Therefore, it is not possible to calculate this parameter precisely for most energetic molecules [8,9,10].
Owens carried out molecular orbital calculations to determine the energy barriers for bond rupture in some energetic molecules [11]. He found that the energy required to break the weakest bond in the molecule correlated well with the impact sensitivity. It has been suggested that the ring-NO2 rupture might be the rate determining step in the initiation of decomposition of many energetic materials [3]. Kuklja [12,13,14] showed by quantum mechanics that cleavage of the N–NO2 bond in RDX requires less energy in an isolated molecule than for a molecule in the bulk of the solid. Interestingly, if the molecule is located near a free surface of the crystal, less energy is required to break the N–NO2 bond than for both the bulk crystal molecule and the isolated molecule. See also references [15,16,17,18,19,20] for other models of sensitivity. Currently it seems that sensitivities are only estimated in terms of general trends, whereas, for instance, relationships to heats of detonation and free space in lattice are also important.
There have been several theoretical characterization studies of 1,2,4-triazoles [21,22,23,24]. Ravi and coworkers [25] have calculated the explosive performance, band gaps, and Mulliken charges of 70 NTO derivatives. The substituted groups were CH3, NO, NO2, and NH2. For NTO derivatives, Turker and Atalar [26] showed that the bond dissociation energy was dependent on the substitution position of the nitro group. When Zbarsky and Yudin [27] nitrated NTO, they found that products with the nitro group attached to nitrogen in the heterocycle were very sensitive. They reported the burn rate of nitro-1,2,4-triazol-5-one with unknown numbers of substituted nitro groups to be 100 mm/s at 10 MPa [27].
Jadhav et al. [28] nitrated both amine nitrogen atoms of NTO and obtained a product (1,3,4-trinitro-1,2,4-triazol-5-one) with an oxygen balance of +21%. They reported an impact sensitivity of 24 J for this compound, indicating that it is less sensitive than RDX, which has a corresponding value of 7.5 J [28]. On the other hand, Dippold et al. [29] found that the nitrated ANTA product 5-nitramino-3-nitro-1,2,4-triazole was more friction- and impact-sensitive than RDX.
In this work, we compute the N-NO2 and C-NO2 bond dissociation energies by using two DFT functionals, B3LYP and M06-2X, and the complete basis set method CBS-4M. We estimate the impact sensitivities of RDX, 2,4,6-trinitrotoluene (TNT), NTO, ANTA, and the nitrated 3-nitro-1,2,4-triazole derivatives. For reference, the Appendix shows the detonation properties and oxygen balance of the molecules. These values are mostly given in reference [30].
Theory and methods
Molecules
Figure 1 shows the molecular structures of RDX, TNT, ANTA and NTO. RDX and TNT are used as reference compounds. The molecular structures of the nitrated NTO and ANTA derivatives are shown in Figs. 2 and 3, respectively. Detonation properties and oxygen balance of the molecules can be found in Table 4 in the Appendix.

Molecular structures of 1,3,5-trinitroperhydro-1,3,5-triazine (RDX), 2,4,6-trinitrotoluene (TNT), 5-amino-3-nitro-1,2,4-triazole (ANTA), and 3-nitro-1,2,4-triazol-5-one (NTO)

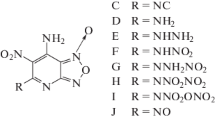
Molecular structures of nitro-substituted NTO compounds. From left to right: 1,3-dinitro-1,2,4-triazol-5-one (1), 2,3-dinitro-1,2,4-triazol-5-one (2), 3,4-dinitro-1,2,4-triazol-5-one (3), and 1,3,4-trinitro-1,2,4-triazol-5-one (4)

Molecular structures of nitro-substituted ANTA compounds. Upper row, from left to right: 5-amino-1,3-dinitro-1,2,4-triazole (5), 5-amino-3,4-dinitro-1,2,4-triazole (6), 5-nitramino-3-nitro-1,2,4-triazole (7). Bottom row, from left to right: 5-nitrimino-3-nitro-1,2,4-triazole (8), 5-nitrimino-1,3-dinitro-1,2,4-triazole (9), 5-nitrimino-3,4-dinitro-1,2,4-triazole (10), 5-nitrimino-1,3,4-trinitro-1,2,4-triazole (11)
Models for impact sensitivity
If the heat of detonation, Q, represents the decomposition energy, the impact sensitivity, I50, can be expressed by Eq. 2.1:
where m is the mass of the drop weight (2.5 kg), g is the gravity constant, a 1 = 0.278331 m, a 2 = 1.1135 × 10−3 m, a 3 = 11.0793 g kcal−1, and a 4 = 1.6606 kcal g−1 [3]. A hybrid model (Eq. 2.2) was also applied, combining the ESP balance parameter and the heat of detonation:
where a 1 = 1.341 × 10−2 m, a 2 = 8.1389, a 3 = 6.7922 g kcal−1 and a 4 = 1.4737 kcal g−1 [3].
Some models are based on the assumption that the two primary factors determining the impact sensitivity are the bond dissociation energy of the weakest C-NO2 or the N–NO2 bond and the energy content of the molecule [19, 31, 42, 43]. The energy content in the models is expressed as the detonation temperature, the total energy of the molecule, and the heat of detonation of the molecule. The models are derived by first assuming that the rate determining step is the elementary unimolecular decomposition.
Computational details
The detonation heat (Q) and temperature (T ex) for the materials were computed in EXPLO5 [43]. For the quantum chemical calculations, we used the DFT or the complete basis set method as implemented in the GAUSSIAN09 software [32]. The bond dissociation energy E b at T = 0 of the bond A–NO2 is
where A(A⋅), E(NO2⋅) and E(A − NO2) denote the ground state electronic energy (open shell model) of the compounds A⋅, NO2⋅, and E(A − NO2), respectively.
Without including the zero point energy, the total energy (E Total) was determined with the hybrid DFT functional B3LYP with the 6-31G(d) basis set. B3LYP has become popular for computation of optimized structures and thermochemical properties for energetic materials. However, all our molecules contain nitro groups, and, according to Brinck and Rahm, neither geometries nor energies have been successfully calculated by B3LYP for such compounds [33]. Yan and coworkers found that this functional gave dissociation energies that were too low for C–N bonds. On average, their calculated values were more than 20 kJ mol−1 lower than the experimental results [34]. For that reason, we included the M06-2X functional in our study. We were inspired by Zhao et al. [35], who reported that this hybrid DFT functional gives better results than B3LYP. In addition to the basis set 6-31G(d), we carried out M06-2X calculations with the larger basis set 6-311+G(2d, p). Our motivation for doing so was that the bond dissociation energies may be dependent on the size of the basis set.
The B3LYP and M06-2X functionals contain a number of constants that were fitted to experimental data. The calculated geometries and energies therefore depend, to some extent, on the parameterization set. Therefore, we also applied a non-DFT method to calculate the bond dissociation energy. This method, CBS-4M, is based on the Hartree-Fock theory and the Møller-Plesset perturbation theory.
Results
The molecular structures were at first geometry optimized with the B3LYP/6-31G (d,p) functional. The optimized structures showed that the NTO and ANTA derivatives had a plane conformation if there were no nitro groups at the neighboring position in the molecule. If a nitro group was at a neighboring position, the nitro groups were twisted out of the plane. None of the optimized geometries had any imaginary frequencies, indicating that these geometries were at least local minima. It can be seen that the isosurface method, as well as the ESP-corrected method, give reasonably good approximations, but both methods underestimate the density of NTO and 4 significantly.
A parameter that may give an indication of the impact sensitivity is the bond dissociation energy. For nitramines, dissociation of the N–NO2 bond is assumed to be the key step in the initiation process [36]. In our bond dissociation energies calculations, the zero point energies were included in order to compare the two DFT functionals with the CBS-4M method. Table 1 shows the N–NO2 bond dissociation energy calculated by the B3LYP/6-31G(d), M06-2X/6-31G(d), M06-2X/6-311+G(2d,p) functionals and the CBS-4M method. Data from Jensen et al. [30] are included in the table. The bond dissociation energies depend on the position of the nitro group in NTO. The N–NO2 bonds of 4 are weak even though the impact sensitivity is measured to be 24 J, and the thermal decomposition, measured by thermogravimetric analysis, started at 194 °C [28].
Weak N–NO2 bonds are also seen in the ANTA derivatives, where the lowest bond dissociation energy was calculated to be 64 kJ mol−1. In general, the N–NO2 bonds in the derivatives were weaker than the N–NO2 bonds in RDX, except for the N–NO2 bond in the nitrimino group. This bond dissociation energy was calculated by M06-2X/6-31G(d) to be around 300 kJ mol−1 which is considerably higher than the N–NO2 bond dissociation energy in RDX.
Some results are not included in Tables 1 and 2 because imaginary frequencies were calculated for the optimized geometries. The bond dissociation energies of C–NO2 were significantly higher than for N–NO2 (Table 2). The C–NO2 bond in the NTO derivatives was weakened when at least one nitro group was substituted to a nitrogen in the 1,2,4-triazole. The same effect was experienced for the ANTA derivatives, but, when a nitro group was substituted to the ANTA NH2 group, the C–NO2 bond dissociation energy was slightly less changed.
If we examine the relationship between the two different DFT functionals and the CBS-4M method, some deviations in the calculated bond dissociation energies are seen. Tables 1 and 2 show that the B3LYP/6-31G(d) method gives a bonding energy that is on average 34 kJ mol−1 lower than calculated by the CBS-4M method. However, the bond dissociation energies for 8, 9, 10 and 11 strongly deviate, as seen in Fig. 4. The bond dissociation energies not correlating are the N–NO2 bonds in the nitrimino functional group.

Bond dissociation energies calculated by B3LYP/6-31G(d) plotted against bond dissociation energies calculated by the CBS-4M method
The way the CBS-4M method and M06-2X/6-31G(d) correlates is displayed in Fig. 5. However, the N–NO2 bond dissociation energy in the nitrimino group in 11 deviates from the trend. By using the larger basis set 6-311+G(2d,p), the bond dissociation energies decreased by 8 kJ mol−1 on average compared to the values obtained by the 6-31G(d) basis set with the M06-2X functional [30]. The B3LYP functional seems to calculate the N–NO2 bond dissociation energy in the nitrimino functional group significantly lower than the M06-2X functional and the CBS-4M method. Since the deviations are relatively large, there is probably an error with the bond dissociation energy computed with the B3LYP functional for the N–NO2 bond in the nitrimino group. Several starting geometries were tested, but all calculations ended up with the same optimized geometries and minimum energies.

Bond dissociation energies calculated by M06-2X/6-31G(d) plotted against bond dissociation energies calculated by the CBS-4M method
Calculated detonation temperature T ex values are given in Table 4 (Appendix) and the E b/T ex ratios are shown graphically in Fig. 6. This figure may thus also give an indication of the impact sensitivities. ANTA and NTO are low-sensitive explosives and exhibit a high E b/T ex ratio compared to those of the considerably more sensitive compounds RDX and 7. Apart from 8, all the NTO and ANTA derivatives have lower E b/T ex ratios than RDX, indicating that they may be more sensitive than RDX.

Calculated E b/T ex ratios for RDX, TNT, NTO, ANTA, and their derivatives 1–11
The impact sensitivities were calculated by the different models and the results are displayed in Table 3. Values of the heat of detonation (Q) are listed in Table 4 (Appendix). Equation 2.1, which is based on the correlation between the impact sensitivity and Q, predicted the low sensitivity of ANTA and NTO fairly well, but there is still a significant disagreement between calculated and measured values. However, this method indicated that 4 might have a sensitivity close to that of TNT, consistent with experimental findings. Equation 2.1 predicted the sensitivity of the derivatives to be at the same level as RDX. Equation 2.2, which is a combination of heat of detonation and the ESP balance parameter, predicted the sensitivity of ANTA and NTO to be higher than the measurement sensitivities. In addition, the impact sensitivity of RDX was by Eq. 2.2 calculated to be 1.2 J, whereas the experimental value is 7.5 J [37].
However, all models indicate that the sensitivities of the ANTA and NTO derivatives are about the same level as that of RDX. The fact that 8 also has a higher E b/T ex ratio than RDX may indicate that the impact sensitivity of this compound is acceptable. Its detonation properties and impulse were also similar to that of RDX. For RDX the Dalton program shows the bond energy of 182 kJ/mol, while the program Psi4 shows 175 kJ/mol when using DFT functional B3LYP and basis sets 6-31G (Dalton) and 6-31G(d) (Psi4).
Calculations showed that the impulse of a mixture of 4 and GAP exceeds 2600 m s−1 [37]. In that respect, 4 is a very interesting compound as a main ingredient in high-energetic compositions. Equations 2.1 and 2.2 show a lower sensitivity than RDX. The measured impact sensitivity is 24 J, indicating that 4 can be handled safely. From this we infer that 4 does not go through the same initial decomposition steps as NTO. It has been proposed that the thermal decomposition of NTO starts with a hydrogen transfer to the nitro group at around 230–260 °C. The next step is either a cleavage of the HONO group or a reorganization of the double bond in NTO, followed by N2 release from the heterocycle [39, 40]. Unlike NTO, 4 does not contain hydrogen atoms, making initiation reactions by hydrogen transfer impossible. This fact may explain the relatively low sensitivity of 4.
Conclusions
Bond dissociation energies were calculated with the DFT functionals B3LYP/6-31G(d) and M06-2X with the 6-31G (d) and 6-311+G(2d,p) basis set, and the non-DFT method CBS-4M was used. These calculations showed that the N–NO2 bonds are weak, and range from 43 kJ mol−1 to 139 kJ mol−1 when calculated at the M06-2X/6-31G(d) level including the zero point energy. One exception is the N–NO2 bond in the nitrimino group, which has a bond dissociation energy as high as 305 kJ mol−1. The N–NO2 nitrimino group bond dissociation energy calculated with the B3LYP functional was between 40 kJ mol−1 and 135 kJ mol−1 lower than the bond dissociation energy calculated by the M06-2X functional or the CBS-4M method. In general, the calculations indicated that most of the derivatives have nearly equal or lower sensitivities than RDX. Previously reported measurements, supported by two of the calculation models indicated that the impact sensitivity of 4 is comparable to, or lower than, that of RDX.
Abbreviations
- [A-NO2]:
-
, Molar concentration
- ANTA:
-
5-amino-3-nitro-1,2,4-triazole
- AP:
-
Ammonium perchlorate
- B3LYP:
-
Hybrid DFT functional
- CBS-4M:
-
Complete basis set method
- D:
-
Detonation velocity
- DFT:
-
Density functional theory
- E a :
-
Activation energy
- E b :
-
Bond dissociation energy
- ESP:
-
Electrostatic potential
- ETotal :
-
Total energy
- GAP:
-
Glycidyl azide polymer
- I 50 :
-
Impact sensitivity
- m :
-
Mass of drop weight
- M06-2X:
-
Hybrid DFT functional
- NTO:
-
3-nitro-1,2,4-triazol-5-one
- P:
-
Detonation pressure
- Q:
-
Heat of detonation
- R:
-
The gas constant, 8.314 J/(mol K)
- RDX:
-
1,3,5-trinitroperhydro-1,3,5-triazine
- T:
-
Temperature
- T ex :
-
Temperature of detonation
- TNT:
-
2,4,6-trinitrotoluene
- V:
-
Volume of detonation products
- ν :
-
Degree of balance between the positive and the negative electrostatic potentials on the isosurface.
References
Politzer P, Murray JS (1996) Relationships between dissociation energies and electrostatic potentials of C–NO2 bonds: application to impact sensitivities. J Mol Struct 376:419–424
Rice BM, Sahu S, Owens FJ (2002) Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J Mol Struct (THEOCHEM) 583:69–72
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106:1770–1783
Ye S, Tonokura K, Koshi M (2003) Energy transfer rates and impact sensitivities of crystalline explosives. Combust Flame 132:240–246
Su-Hong G, Cheng X-L, Wu L-S, Yang X-D (2007) Correlation between normal mode vibrations and impact sensitivities of some secondary explosives. J Mol Struct (THEOCHEM) 809:55–60
Tan B, Long X, Peng R, Li H, Jin B, Chu S, Dong H (2010) Two important factors influencing shock sensitivity of nitro compounds: bond dissociation energy of X-NO2 (X= C,N,O) and Mulliken charges of nitro group. J Hazard Mater 183:908–912
Zhu W, Xiao H (2010) First-principle band gap criterion for impact sensitivity of energetic crystals: a review. Struct Chem 21:657–665
Yan Q-L, Zeman S (2013) Theoretical evaluation of sensitivity and thermal stability for high explosives based on quantum chemistry methods: a brief review. Int J Quantum Chem 113:1049–1061
Chen Z-X, Xiao H-M (2014) Quantum chemistry derived criteria for impact sensitivity. Propellants Explos Pyrotech 39:487–495
Politzer P, Murray JS (2014) Detonation performance and sensitivity: a quest for balance. Adv Quantum Chem :1–29
Owens FJ (1996) Calculation of energy barriers for bond rupture in some energetic materials. J Mol Struct (THEOCHEM) 370:11–16
Kuklja MM (2001) Chemical decomposition of solid cyclotrimethylene trinitramine. Comput Nanosci 2001:73–76
Sharia O, Kuklja MM (2012) Rapid materials degradation induced by surface and voids: ab initio modeling of β-octotetramethylene tetranitramine. J Am Chem Soc 134:11815–11820
Kuklja MM (2014) Quantum-chemical modeling of energetic materials: chemical reactions triggered by defects. Deformations, and electronic excitations. Adv Quantum Chem 69:71–146
Storm CB, Stine JR, Kramer JF (1990) In: Bulusu SN (ed) Chemistry and physics of energetic materials—sensitivity relationships in energetic materials. Kluwer, Dordrecht, p 605
Fried LE, Ruggiero AJ (1994) Energy transfer rates in primary, secondary, and insensitive explosives. J Phys Chem 98:9786–9791
Pospisil M, Vavra P, Concha MC, Murray JS, Politzer P (2010) A possible crystal volume factor in the impact sensitivities of some energetic compounds. J Mol Model 16:895–901
Politzer P, Murray JS (2014) Impact sensitivity and crystal lattice compressibility/free space. J Mol Model 20:2223
Fried LE, Manaa MR, Pagoria PF, Simpson RL (2001) Design and synthesis of energetic materials. Ann Rev Mater Sci 31:291–321
Politzer P, Murray JS (2015) Impact sensitivity and the maximum heat of detonation. J Mol Mod :262. doi: 10.1007/s00894-015-2793-z
Rui-Zhou Z, Xiao-Hong L, Xiang-Zhou Z (2012) Theoretical studies on a series of 1,2,4-triazoles derivatives as potential high energy density compounds. J Chem Sci 124:995–1006
Ravi P, Tewari SP (2013) A DFT study of tautomers of 3-amino-1-nitroso-4-nitrotriazol-5-one-2-oxide. J Mol Model 19:2539–2547
Wu Q, Zhu W, Xiao H (2014) Quantum chemical studies on three novel 1,2,4-triazole N-oxides as potential insensitive high explosives. J Mol Model 20:2441
De Paz JLG, Ciller J (1994) Structure and tautomerism of ANTA (aminonitrotriazole). Propellants Explos Pyrotech 19:32–41
Ravi P, Babu BK, Tewari SP (2013) Theoretical investigations on the structure, density thermodynamics and performance properties of amino-, methyl-, nitroso- and nitrotriazolones. J Mol Model 19:33–48
Turker L, Atalar T (2006) Quantum chemical study on 5-nitro-2,4-dihydro-3H-1,2,4-triazol-3-one (NTO) and some of its constitutional isomers. J Hazard Mater A137:1333–1344
Zbarsky VL, Yudin NV (2005) Kinetics of the synthesis of NTO in nitric acid. Propellants Explos Pyrotech 30:298–302
Jadhav HS, Talawar MB, Dhavale DD, Asthana SN, Krishnamurthy VN (2005) Synthesis, characterization and thermolysis of 2,4-dihydro-2,4,5-trinitro-3H-1,2,4-triazol-3-one (DTNTO): a new derivative of 3-nitro-1,2,4-triazol-5-one (NTO). Indian J Eng Mater Sci 12:467–471
Dippold AA, Klapötke TM, Martin FA, Wiedbrauk S (2012) Nitraminoazoles based on ANTA—a comprehensive study of structural and energetic properties Eur J Inorg Chem:2429–2443
Jensen TL, Moxnes JF, Kjønstad EF, Unneberg E (2016) A study of the detonation properties, propellant impulses, impact sensitivities and synthesis of nitrated ANTA and NTO derivatives. Cent Eur J Energ Mater 13:445–467
Mathieu D (2012) Theoretical shock sensitivity index for explosives. J Phys Chem A 116:1794–1800
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford
Brinck T, Rahm M (2014) In: Brinck T (ed) Green energetic materials, Chap. 2. Wiley, Chichester, p 17
Yao X-Q, Hou X-J, Jiao H, Xiang H-W, Li Y-W (2003) Accurate calculations of bond dissociation enthalpies with density functional methods. J Phys Chem A 107:9991–9996
Zhao Y, Truhlar DG (2008) How well can new-generation density Functionals describe the Energetics of bond-dissociation reactions producing radicals. J Phys Chem A 112:1095–1099
Klapötke TM (2011) Chemistry of high energy materials. de Gruyter, Berlin,
Meyer R, Köhler J, Homborg A (2007) Explosives, 6th edn. Wiley-VCH, Weinheim,
Lee KY, Storm CB, Hiskey MA, Coburn MD (1991) An improved synthesis of 5-amino-3-nitro-1H-1,2,4-triazole (ANTA), a useful intermediate for the preparation of insensitive high explosive. J Energ Mater 9:415–428
Oxley JC, Smith JL, Zhou ZL, Mckenny RL (1995) Thermal decomposition studies on NTO and NTO/TNT. J Phys Chem 99:10383–10391
Sinditskii VP, Smirnov SP, Egorshev VY (2007) Thermal decomposition of NTO: an explanation of the high activation energy. Propellants Explos Pyrotech 32:277–287
Song X, Cheng X, Yang X, Li D, Linghu R (2008) Correlation between the bond dissociation energies and impact sensitivities in nitramine and polynitro benzoate molecules with polynitro alkyl groupings. J Hazard Mater 150:317–321
Mathieu D (2013) Towards a physically based quantitative modeling of impact sensitivities. J Phys Chem A 117:2253–2259
Suceska M (2010) EXPLO5, version 5.04. Zagreb
Acknowledgment
We thank Tor Erik Kristensen at the Norwegian Defense Research Establishment for interesting discussions during the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Rights and permissions
About this article

Cite this article
Moxnes, J.F., Frøyland, Ø. & Risdal, T. A computational study of ANTA and NTO derivatives. J Mol Model 23, 240 (2017). https://doi.org/10.1007/s00894-017-3408-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3408-7