
Abstract
Density functional theory calculations at B3LYP/6-31G** and B3P86/6-31G** levels were performed to predict the densities (ρ), detonation velocities (D), pressures (P) and the thermal stabilities for a series of 1,2,4-triazole derivatives for looking high energy density compounds (HEDCs). The heats of formation (HOFs) are also calculated via designed isodesmic reactions. The calculations on the bond dissociation energies (BDEs) indicate that the position of the subsitutent group has great effect on the BDE and the BDEs of the initial scission step are between 31 and 65 kcal/mol. In addition, the condensed phase heats of formation are also calculated for the title compounds. These results would provide basic information for further studies of HEDCs.

Densities, detonation velocities and pressures for a series of 1,2,4-triazole derivatives, as well as their thermal stabilities, were investigated to look for high energy density compounds (HEDCs). Heats of formation (HOFs) were also calculated via designed isodesmic reactions. 5,5′-Dinitro-3,3′-bi-1,2,4-triazole, 3-nitro-1-picryl-1,2,4-triazole and 4-(2,4-dinitrobenzyl)-3,5-dinitro-1,2,4-triazole satisfy the quantitative standard of HEDC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Energetic materials (explosives, propellants and pyrotechnics) are used extensively both for civil and military applications. Continuous strong efforts have been made to develop new materials having good thermal stability, impact and shock insensitivity, better performance, economic and environmentally friendly syntheses in order to meet the requirements of future military and space applications.[1, 2] Explosives containing triazole rings have the advantages of less molecular weight, high nitrogen content and density, good thermal stability, low impact sensitivity and large explosive volume, so that they can be used as outstanding explosives. Triazoles and their derivatives have also been widely used in the field of agriculture, biology, medicine and photoelectricity duplication.[3–5]
The nitro group is an important group for energetic materials.[6] Through increasing numbers of the nitro group, the compounds’ density and the number of mole gaseous combustion products formed by per gram of material can be increased, thereby enhancing propellant performance. The nitrotriazole derivatives are interesting energetic compounds.[7] The syntheses of nitrotriazole derivatives as energetic materials and as intermediates to energetic materials have received a great deal of attention in the past 10 years.[8] Krishna Kumar et al.[9] have investigated the hydrogen bonding and molecular vibration of 3,5-diamino-1,2,4-triazole. Karayel et al.[10] reported the synthesis of 4-(2-phenylethyl)-5-(2-furyl)-2, 4-dihydro-3H- 1,2,4- triazole-3-thione by X-ray diffraction and molecular modelling techniques. Krishnakumar et al.[11] also reported the synthesis of 3-mercapto-1,2,4-triazole by using polyethylene pellet technique.
Typical triazoles, such as 3-nitro-1,2,4-riazol-5-one (NTO) and 5-amino-3-nitro-1,2,4-triazole (ANTA), have also been investigated experimentally and theoretically.[12–14] Prabhakaran et al.[15] have evaluated the kinetic parameters of NTO using various kinetic models. Nitrotriazole derivatives represent a new generation of energetic materials, which are of interest due to their potential high density, energy and properties as solid propellant oxidizers.[16] Exploring microscopic pyrolysis mechanism, which addresses the question of how an important impulse can initiate rapid exothermic reactions leading to the detonation of explosive solids, is always the research target of both theoretical and experimental chemists.
In general, the initiation of detonation involves a complex interplay of molecular, crystalline and physical factor.[17–19] Kamlet and Adolph thought that correlations with a single factor can indeed exist if others are kept as uniform as possible.[17, 18] However, Brill and James[19] pointed out that a correlation does not necessarily imply a causal relationship; it may simply be symptomatic.
It is well-known that the dissociation of the weakest bond of an explosive molecule has been expected to play an important role in the initiation of detonation. Researches showed that R–NO2 (R–C, N or O) bond is the weakest bond in energetic ring molecules and the rupture of this bond is the first step in decomposition process.[20–22] For a compound containing several nitro groups, it is necessary to determine which bond is the weakest bond.
Heat of formation (HOF) is one of the most crucial thermodynamic quantities. It is required to estimate the amount of energy released or absorbed in a chemical reaction, to calculate other thermodynamic functions and, what is more important, to assess the stability of a molecule.[23–27] However, to the best of our knowledge, less experimental BDE values are available, and the weakest bond has not been identified and studied for the1,2,4-triazole derivatives. For HOFs, the values for the title compounds are at present uncertain. Further, because less than 0.02% of known organic species have had their HOFs measured,[28] the application of quantum methods is both inevitable and desirable, provided that reasonable accuracy can be obtained.
In this paper, the HOFs have been calculated for several 1,2,4-triazole derivatives using density functional theory B3LYP and B3P86 methods with 6-31G** basis set via designed isodesmic reactions. Thermal stability was evaluated via BDE. Results from different methods were compared. The condensed phase HOFs and detonation performance data were also calculated for the studied compounds. These results provide useful information for the molecular design of novel HEDMs.
2 Theory and computational details
The density functional theory (DFT)[29, 30] has been used to evaluate BDEs and HOFs of interested molecules. Geometry optimizations, energy and frequency calculations were carried out with the Gaussian03 package[31] for nitrotriazole derivatives. All calculations of molecular geometry and energy were performed using DFT method, Becke 3 parameters exchange and Lee, Yang and Parr correlation functionals[32, 33] and Perdew’s 86(P86),[34] with the default Gaussian convergence criteria. 6-31G** basis set is used.
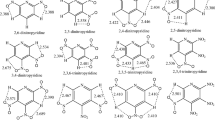
The studied nitroiazole derivatives are 5,5′-Dinitro-3,3′-bi-1,2,4-triazole (C4H2N8O4), 3-Nitro-1-picryl-1,2,4-triazole(C8H3N7O8), 3-amino-5-picrylamino-1,2,4 -triazole(C8H6N8O6), ammonium-3,5 -dinitro-1,2,4-triazolate(C2H4N6O4), 4-Methyl-3,5-dinitro-1,2,4-triazole(C3H3N5O4), 4-(2-nitroethyl)-3,5-dinitro-1,2,4-triazole(C4H4N6O6), 4-picrylamino-1,2,4-triazole(C8H5N7O6), 4-(2,4-dinitrobenzyl)-3,5-dinitro-1,2,4-triazole (C9H5N7O8). Here, we assume a homolytic cleavage of C–NO2 bond while calculating BDEs, the fragments are radical species. The calculations of geometry and energy for all fragments were performed using the spin-unrestricted method with the same basis set 6-31G**. Vibrational analysis has also been performed for each stationary point to verify a minimum energy structure and to provide zero-point energies (ZPEs) and thermal correction. The < S 2 > values are all very close to 0.75, which shows negligible spin contamination of pure doublets states for fragment open-shell systems. For every molecule, we optimized several possible stereoisomers, and selected the structure with the lowest energy as the most stable structure. Figure 2 shows the molecular frameworks of the studied title compounds.

Molecular frameworks and chemical names.
The R–NO2 bond strength is obtained by calculating the BDE, defined here as the difference between the total energy of the parent molecule and the energies of the products of the unimolecular dissociation.[35] For example, for 4–Picrylamino-1,2,4-triazole (C8H5N7O6), the BDE is
The bond dissociation energy with ZPE correction
where ΔZPE is the difference between the zero-point energies (ZPE) of the products and the reactants.
The predictions of HOFs adopt the hybrid DFT B3LYP and B3P86 methods with 6-31G** basis set via designed isodesmic reactions.[36] The method of isodesmic reactions has been employed very successfully to calculate HOFs.[37–39] The HOFs for the title compounds were derived from the following isodesmic reactions.








For the isodesmic reaction (3)–(10), heat of reaction ΔH 298 at 298 K can be calculated from the following equation
where ΔH f,P and ΔH f,R are the HOFs of products and reactants at 298 K, respectively. The HOFs of the title compounds can be figured out when the heat of reaction is known. The HOFs at 298.15 K can be calculated from the following equation
where ΔE 0 and ΔZPE are the total energy difference and the zero-point energy difference between products and reactants at 0 K, respectively; ΔH T is the changes in thermal correction to enthalpies between products and reactants; \(\sum\limits_{\it product} {\Delta H_f^0 } \) and \(\sum\limits_{\it reactant} {\Delta H_f^0 } \) are sums of the heats of formation for products and reactants in gas at 298.15 K, respectively. Δ(PV) equals ΔnRT for reaction in gas phase.
3 Results and discussions
3.1 The bond dissociation energies of several 1,2,4-triazole derivatives
The thermal stability of title compounds should be emphasized because of high energetic insensitive explosive. Natural bond orbital analysis can be used to investigate the relative stability of molecules,[40] while the stability of a molecule is usually evaluated by its bond dissociation energy, especially for energetic materials. So, in this paper, we calculated the dissociation energies for the possible initial steps in the pyrolysis route. It should be pointed out that we select the C–NO2, N–NH2, N–CH3 and C–NH2 bonds as the possible breaking bond at the B3LYP/6-31G** and B3P86/6-31G** levels. The values of BDEs are listed in table 1. As has been suggested by Chung et al.,[41] a molecule should have more than 20 kcal/mol barrier to dissociate in order to be considered as a viable candidate for new HEDMs. From table 1, we can conclude that all the molecules investigated are all viable candidates for new HEDMs. From table 1, it is also noted that the BDE calculated by B3P86 functional is about 4.2 kcal/mol higher than the result calculated by B3LYP functional, which is consistent with the result calculated before.[42]
For compound (1), the BDEs of the C1–N1 and C\(_{1\hbox{'}}\)–N\(_{1\hbox{'}}\) bonds are equivalent and the BDE of the C1–N1 bond is 60.1 kcal/mol. For compound (2), the BDEs of the C2–N2 and C6–N6 bonds are equivalent, so there are three possible breaking bonds, the C1–N1, C2–N2, and C4–N4 bonds. From table 1, we can see that the C2–N2 BDE is the smallest and is equal to 44.9 kcal/mol for B3LYP/6-31G** method. This means that the C2–N2 bond may be the initial scission step.
The BDEs of the C2–N2 and C6–N6 bonds are equivalent for compound (3); the C2–N2 BDE is 54.5 kcal/mol for B3LYP method, which is smaller than the C4–N4 and C–NH2 BDEs. This means that the C2–N2 bond is the possible initial scission step. For compound (4),the BDE of the N2–N3 bond is 50.4 kcal/mol, which is larger than that of the C5–N5bond and smaller than that of the C1–N1 bond.
For compound (5), the BDEs of the C1–N1 and C5–N5 bonds are equivalent and are all equal to 60.9 kcal/mol, while the BDE of the C2–N2 bond is 87.4 kcal/mol. The BDEs of the C1–N1 and C5–N5 bonds are also equivalent for compound (6) and is equal to 61.3 kcal/mol, which is larger than that of the C3–N3 bond. For compound (8), the BDEs of the C1–N1 and C5–N5 bonds are equivalent and are all equal to 58.8 kcal/mol, which are smaller than the BDEs of C2–N2 and C4–N4 bonds. When compared the structures of compounds (5), (6) and (8), it is noted that the substituted group has great effect on the BDE of the weakest bond. The methyl group at 4-position on the 1,2,4-triazole ring is substituted by 2-nitroethyl group, the BDE of the weakest bond decreases from 60.9 kcal/mol to 51.3 kcal/mol. When the 2-nitroethyl group at 4-position on the 1,2,4-triazole ring is substituted by 2,4-dinitrobenzy group, the BDE of the weakest bond increases from 51.3 kcal/mol to 58.8 kcal/mol. In addition, it is also noted that the BDE of the weakest bond is 51.3 kcal/mol when the substituted group is the 2-nitroethyl group at 4-position on the 1,2,4-triazole ring, while the BDE of the weakest bond drastically decreases to 31.9 kcal/mol when the substituted group is the ammonium group at 1-position on the 1,2,4-triazole ring.
For compound (7), the BDEs of the C2--N2 and C6--N6 bonds are equivalent and are equal to 53.4 kcal/mol, which is smaller than that of the C4--N4 bond and may be the initial scission step.
For the title compounds, the BDE values of the initial scission step are between 31 and 65 kcal/mol, which are higher than those of piperidine and diazocine compounds[43] and polynitro benzoate molecules.[44]
From the above analysis, it is noted that compound (4) is the most reactive compound, while compound (5) is the least reactive compound for the 1,2,4-triazole derivatives studied in our paper.
In addition, the position of the substitutent group has great effect on the BDE. The eight nitrotriazole compounds have many isomeric compounds. Here, we calculated some isomeric structures of the compound (5) with B3LYP/6-31G** method and obtained the dissociation energies for the possible initial steps in the pyrolysis route. The results are listed in table 2. The molecular structures of six compounds are listed in figure 2.

Molecular frameworks and chemical names of some isomeric structure of compound (5) (4-Methyl-3,5-dinitro-1,2,4-triazole).
From table 2, it is noted that the position of nitro group has an important effect on the BDE for the molecules with all nitro group attached to C atoms. For example, the BDE of C--NO2 on C1 site is 1.9 kcal/mol lower than the BDE of C--NO2 on C5 site for 2-methyl-3,5-dinitro-1,2,4-triazole and the BDE of the weakest bond is 62.5 kcal/mol. For 1-methyl-3,5-dinitro-1,2,4-triazole, the BDE of C--NO2 on C1 site is 1.9 kcal/mol larger than the BDE of C--NO2 on C5 site and the BDE of the weakest bond is 61.7 kcal/mol. The BDEs of C1--N1 and C5--N5 bonds are the same and equal to 60.9 kcal/mol for compound (5), which are the weakest bonds. A possible explanation for the above behaviour is that the presence of the weak interaction between O atom of nitro group and adjacent hydrogen atom, weakens the C--NO2 bond length. In addition, this indicates that the compound is more reactive when the methyl group is attached to the nitrogen atom on 1-position of 1,2,4-triazole.
From table 2, it is also noted that N--NO2 bond strength is between 20 and 35 kcal/mol for the molecules with nitro group attached N atom. And N--NO2 bond is the weakest bond for the molecules containing N--NO2 linkage (such as 5-methyl-3,4-dinitro-1,2,4-triazole, 3-methyl-2,5-dinitro-1,2,4-triazole, 5-methyl-2,3- dinitro-1,2,4-triazole). This indicates that the rupture of N--NO2 bond is the initial site in the decomposition process.
3.2 Heats of formation
Table 3 lists the total energies and zero-point energies at the B3LYP/6-31G** method for several reference compounds involved in the isodesmic reactions (3)--(10). Thermodynamic information was obtained from scaled vibrational frequencies with scaling factors taken from Scott and Radom.[45] The experimental HOFs of reference compounds CH4, C2H6, CH3NO2, CH3NH2, C6H6 are taken from refs.[46, 47]. Table 4 shows the total energies, zero-point energies, the values of the thermal corrections for title compounds, and the values of HOFs obtained via Eq. (12). Previous studies showed that theoretically predicted values of HOFs were in good agreement with experiments by choosing appropriate reference compounds in the isodesmic reactions.[38–44]
It is noted from table 4 that the HOFs calculated by B3LYP/6-31G** and B3P86/6-31G** methods are similar and the HOFs calculated by B3P86/6-31G** method are slightly smaller than those by B3LYP/6-31G** method. From the calculated results, we can see that substituent groups greatly affect the HOFs of the title compounds. The gas phase HOF of 1,2,4-triazole is 46.1 kcal/mol,[44] while the HOF of compound (1) is 95.75 kcal/mol at B3LYP/6-31G** level. This shows that the HOF of the compound drastically increases when a H group at 5-position is substituted by nitro group and a H group at 3-position is substituted by 5-nitro-1,2,4-triazole for 1,2,4-triazole. The HOFs of compounds (5), (6) and (8) are 46.99, 37.61 and 60.23 kcal/mol, respectively. This shows that the HOF increases when a 2-nitroethyl group at 4-position on 1,2,4-triazole ring is substituted by a methyl group, while the HOF decreases when the 2,4-Dinitrobenzyl group at 4-position on 1,2,4-triazole ring is substituted by a methyl group. The HOF of compound (7) is 81.37 kcal/mol. When the H atom at 3-position of compound (7) is substituted by an amino group and the picrylamino group at compound (7) moves from 4-position to 5-position, the HOF of the compound obtained decreases drastically and is equal to 50.48 kcal/mol.
In view of these theoretical results, compound (2) has smaller initial scission bond and bigger HOFs, so it is advisable for us to select compound (2) in order to obtain the desirable energetic materials possessing high explosive performance.
From table 4, it can be seen that the calculated HOFs for our investigated molecules are all endothermic, which is desirable for HEDMs. Since no experimental values are available, we have also calculated the HOFs at B3P86/6-31G** level. Comparing the calculated HOFs by the two different levels, one can obtain that the discrepancy of the two levels is very small, with deviations ranging from 0 to 4.0 kcal/mol and with an average value of 1.3 kcal/mol. It is noted that the discrepancy of the two levels for compound (4) is about 3.9 kcal/mol. The linear relationship between the HOFs obtained by B3LYP/6-31G** level and B3P86/6-31G** level is very good:
with R2 = 0.998. The coherency obtained from the two levels demonstrates precision and suggests reasonable accuracy.
The condensed phase \(\Delta H_f^0 \) is the potential performance of the energetic material of interest. In order to accommodate this need, we compute the condensed phase \(\Delta H_f^0 \) of all mentioned energetic compounds through the same method of Byrd and Rice. According to Hess's law, the condensed phase heat of formation \(\left( {\Delta H_{f\left( c \right)}^0 } \right)\) can be obtained by
where \(\Delta H_{f\left( g \right)}^0 \) is the predicted gas phase heat of formation, \(\Delta H_{\it sub}\) is the heat of sublimation. The heat of sublimation can be represented as
where the constants a, b, and c are determined through a least-squares fit of Eq. (15). SA is the surface area of the 0.001 electron/bohr3 isosurface of the electron density of the molecule, \(\sigma_{\it tot}^2 \) is a measure of the variability of electronic potential on the surface, and v is the degree of balance between the positive and negative charges on the isosurface. The latter two quantities have been shown by Politzer et al. to be important in treating macroscopic properties that are dependent on non-covalent electrostatic interactions.[48–50]
The B3LYP hybrid generalized-gradient approximation (GGA) density functional theory was used with 6-311+ +G(2df,2p) basis set to optimize geometries and determine the densities for generating the electrostatic potentials (ESPs) and the atom and group equivalents. The optimized structure is assumed to correspond to a local potential energy minimum. The computed condensed phase heats of formation are listed in table 5.
3.3 Detonation performance data
Detonation velocity (D) and pressure (P) are the most important characteristics of energetic materials. For a series of the explosives with CHNO elements, detonation velocities and pressures can be calculated by using the Kamlet--Jacobs equation[51, 52]:
where each term in Eqs. 16 and 17 is defined as follows: P, detonation pressure (GPa); D, the detonation velocity (km/s); ρ, the packed density (g/cm3); Φ, the characteristics value of explosives; N, the moles of gas produced by per gram of explosives; \(\overline M \), an average molar weight of detonation products; and Q, the estimated heat of detonation (KJ/g). Here, the parameters N, \(\overline M \), and Q were calculated according to the chemical composition of each explosive[51, 52] as listed in table 6. The HOFs calculated by B3LYP/6-31G** level are used. The density of each compound was predicted according to the reference,[53] in which the electrostatic potential is considered.
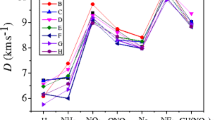
Table 7 collects the predicted V, ρ, Q, D and P of the title compounds. The experimental data[54] of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) and 1,3,5,7-tetranitro-1,3,5,7- tetraazacyclooctane (HMX) are also listed in table 7. It is noted that the values of ρ, D, P for compound (1) are 1.85 g/cm3, 9.72 km/s and 48.2GPa, respectively, which are bigger than those for RDX. For compound (2), the calculated detonation velocity is 9.17 km/s, which is larger than those of RDX and HMX, and the calculated detonation pressure is 42.0 Gpa, which is greater than to that of RDX and HMX. The detonation velocity and detonation pressure for compound (8) are 9.43 km/s and 45.8 GPa, respectively, which are also higher than those for RDX and HMX. Therefore, we think that the three compounds satisfy the quantitative standard of high energetic density compound (HEDC) (ρ ≈ 1.9 g/cm3, D ≈ 9.0 km/s, P = 40 GPa).[55]
The detonation properties of compounds (6), (5) and (3) are not as large as those of HMX and RDX, 7.53 km/s and 22.5 Gpa for compound (6), 7.87 km/s and 25.9 Gpa for compound (5), 8.16 km/s and 31.3 Gpa for compound (3). The small HOF value brings the low detonation velocity and pressure.
4 Conclusions
Using density functional theory, we have calculated the thermochemistry properties such as BDEs, HOFs for the 1,2,4-triazoles derivatives. We have obtained the initial scission bond for the title compounds through calculating the possible initial steps in the pyrolysis route. The results show that compound (4) is the most reactive compound, while compound (5) is the least reactive compound for the 1,2,4-triazole derivatives studied in this work. HOFs at 298 K in gas are calculated via isodesmic reactions at two different calculation levels B3LYP/6-31G** and B3P86/6-31G**. The coherency of the HOFs obtained from the two levels demonstrated our calculational accuracy. The condensed phase heats of formation and detonation performance data of the title compounds are also calculated according to the HOFs calculated by B3LYP/6-31G** level. Compounds (1), (2) and (8) can be considered for HEDC because of the excellent detonation properties.
References
Sikder A K and Sikder N 2004 J. Hazard. Mater. 112 1
Sikder A K, Maddalla G, Agraval J P and Singh H 2001 J. Hazard. Mater. 84 1
Olcay B and Hakan B 2006 Molecules 11 469
Thomas S, Biswas N, Venkateswaran S, Kapoor S, D’Cunha R and Mukherjee T 2005 Chem. Phys. Lett. 402 361
Chen L-M, Chen J-C, Luo H, Liao S-Y and Zheng K-C 2011 J. Theor. Comput. Chem. 10 581
Fried L E, Manaa M R, Pagoria P F and Simpson R L 2001 Annu. Rev. Mater. Res. 31 291
Boyer J S 1986 Nitrozoles (Deerfield Beach: VCH Publishers) Vol. 1
Yuxiang O, Boren C, Jiarong L, Shuan D, Jianjuan L and Huiping J 1994 Heterocycles 38 1651
Krishna Kumar V, Keresztury G, Sundius T and John Xavier R 2005 Spectrochim. Acta A 61 261
Karayel A and Ozbey S 2008 Struct. Chem. 19 391
Krishnakumar V and John Xavier R 2004 Spectrochim. Acta A 60 709
Azhary EI A A, Suter H U and Kubelka J 1998 J. Phys. Chem. A 102 620
Fang G Y, Xu L N, Xiao H M and Ju X H 2005 Acta Chimica Sinica 63 1055
Xu L N, Xiao H M, Fang G Y and Ju X H 2005 Acta Chimica Sinica 63 1062
Prabharan K V, Naidu S R and Kurian E M 1994 XRD, Thermochim. Acta 241 199
Sikder A K, Geetha M, Sarwade D B and Agrawal J P 2001 J. Hazard. Mater. 82 1
Kamlet M J 1976 in Proceedings of the 6th Symposium (International) Deton. Report No. ACR 221 (Office of Naval Research), p. 312
Kamlet M J and Adolph H G 1979 Propell. Expl. 4 30
Brill T B and James K 1993 Chem. Rev. 93 2667
Murray J S and Politzer P 1990 Structure–sensitivity relationships in energetic compounds, in: Bulusu S N (Ed.), Chemistry and Physics of Energetic Materials (Netherlands: Kluwer Academic Publishers), p. 157
Zhang S W and Truong T N 2000 J. Phys. Chem. A 104 7304
Zhao Q, Zhano S and Li Q S 2005 Chem. Phys. Lett. 407 105
Ju X H, Li Y M and Xiao H M 2005 J. Phys. Chem. A 109 934
Fan X W and Ju X H 2008 J. Comput. Chem. 29 505
Rice B M, Pai A V and Hare J 1999 Combust. Flame 118 445
Cobos C J 2005 J. Mol. Struct. (THEOCHEM) 714 147
Fan X W, Ju X H, Xiao HM and Qiu L 2006 J. Mol. Struct. (THEOCHEM) 801 55
Luo Y R 2003 Handbook of bond dissociation energies in organic compounds (Boca Raton, FL: CRC Press)
Parr R G and Yang W 1989 Density functional theory of atoms and molecules (Oxford: Oxford University Press)
Seminario J M and Politzer P 1995 Modern density functional theory: A tool for chemistry (Amsterdam: Elsevier)
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Zakrzewski V G, Montgomery J A, Stratmann R E, Burant J C, Dapprich S, Millam J M, Daniels A D, Kudin K N, Strain M C, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson G A, Ayala P Y, Cui Q, Morokuma K, Malick D K, Rabuck A D, Raghavachari K, Foresman J B, Cioslowski J, Ortiz J V, Baboul A G, Stefanov B B, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin R L, Fox D J, Keith T, Al-Laham M A, Peng C Y, Nanayakkara A, Gonzalez C, Challacombe M, Gill P M W, Johnson B, Chen W, Wong M W, Andres J L, Gonzalez C, Head Gordon M, Replogle E S and Pople J A 2003 GAUSSIAN 03, Revision B.02 (Pittsburgh PA: Gaussian Inc.)
Becke A D 1993 J. Chem. Phys. 98 5648
Lee C, Yang W and Parr RG 1988 Phys. Rev. B 37 785
Perdew J P 1986 Phys. Rev. B 33 8822
Rice B M, Sahu S and Owens F J 2002 J. Mol. Struct. (Theochem) 583 69
Hahre W J, Ditchfield R, Radom L and Pople J A 1970 J. Am. Chem. Soc. 92 4796
Li X-H, Zhang R-Z, Yang X-D and Zhang H 2007 J. Mol. Struct. (Theochem) 815 151
Li X-H, Zhang R-Z, Cheng X-L and Yang X-D 2007 J. Theor. Comput. Chem. 6 449
Li X-H, Zhang R-Z, Zhang X-Z, Cheng X-L and Yang X-D 2007 J. Theor. Comput. Chem. 6 675
Leena R, Jissy A K, Krishnapillai Girish K and Ayan D 2011 J. Phys. Chem. C 115 21858
Chung G S, Schimidt M W and Gordon M S 2000 J. Phys. Chem. A 104 5647
Li X-H, Zhang R-Z, Zhang X-Z, Yang X-D and Cheng X-L 2007 Chinese J. Struct. Chem. 26 1481
Fan X-W, Ju X-H and Xiao H-M 2008 J. Hazard. Mater. 156 342
Song X-S, Cheng X-L, Yang X-D, Li D-H and Rongfeng L-H 2008 J. Hazard. Mater. 150 317
Scott A P and Radom L 1996 J. Phys. Chem. 100 16502
Hahre W J, Radom L and Schleyer P V R 1986 Ab initio molecular orbital theory (New York: Wiley)
Frenkel M, Kabo G J, Marsh K N, Roganov G N and Wilhoit C R 1994 Thermodynamics of organic compounds in the gas state (College Station, TX: Thermodynamic Research Center) Vol. II
Politzer P, Murray J S, Brinck T and Lane P 1994 In immunoanalysis of agrochemicals (Washington, DC: American Chemical Society)
Murray J S and Politzer P 1994 Quantitative treatment of solute/solvent interactions (Amsterdam: Elsevier Scientific)
Politzer P and Murray J S 1998 J. Phys. Chem. A 102 1018
Kamlet M J and Jacobs S J 1968 J. Chem. Phys. 48 23
Zhang X-H and Yun Z-H 1989 Explosive chemistry (Beijing, Peoples Republic of China: National Defense Industry Press)
Politzer P, Martinez J, Murray J S, Concha M C and Toro-Labbe A 2009 Molecul. Phys. 107 2095
Ou Y X and Chen J J 2005 The high energy and density compounds, 1st ed. (Beijing: National Defense Industry Press)
Xiao H M, Xu X J and Qiu L 2008 Theoretical design of high energy density materials (Beijing: Science Press)
Acknowledgements
We thank the National Natural Science Foundation of China (Grant 10774039) and the grant from Development Program in Science and Technology of Henan Province (No. 112300410206), Scientific and Technical Research Foundation for the Education Department of Henan Province (No. 12A140004) for their support to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rui-Zhou, Z., Xiao-Hong, L. & Xian-Zhou, Z. Theoretical studies on a series of 1,2,4-triazoles derivatives as potential high energy density compounds. J Chem Sci 124, 995–1006 (2012). https://doi.org/10.1007/s12039-012-0304-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-012-0304-7