
Abstract
There is considerable evidence, which we discuss, indicating that compressibility and available free space in the crystal lattice are among the factors that govern the sensitivity of an explosive compound. Expanding and extending earlier work, we demonstrate, for 25 explosives, that there is an overall general tendency for greater impact sensitivity as the estimated free space per molecule increases. More specific relationships can be discerned by looking at subgroups of the compounds. The nitramine sensitivities, most of which are quite high, increase nearly linearly but only very gradually with free space. The nitroaromatics cover a wide range of sensitivities but all have an approximately similar intermediate level of free space. The remaining types of compounds show a reasonable sensitivity–free space relationship with one outlier: FOX-7 (1,1-diamino-2,2-dinitroethylene).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Sensitivity
In the context of energetic materials, such as explosives and propellants, the term “sensitivity” refers to the vulnerability of a material to unintended detonation due to an accidental stimulus; this could be impact, shock, electrical sparks, etc. [1–6]. Minimizing this vulnerability while yet attaining a high level of detonation performance (when intended) are very important but somewhat conflicting objectives in developing new explosive formulations.
Impact sensitivity, which is what is most commonly measured, depends upon not only molecular and crystalline factors but also upon environmental conditions and the physical state of the material: the sizes and shapes of the crystals, their hardness and roughness, etc. [7–11]. Achieving reasonably reproducible measured impact sensitivities is accordingly difficult. Great care is taken to employ very specific and uniform procedures and conditions in preparing and testing the materials [2, 8, 9, 12–14]; however impact sensitivity measurements should be viewed as providing only “crude, qualitative” estimates [12]. They are nevertheless extremely important from the standpoints of (a) using the materials, and (b) learning what factors govern sensitivity, and how it can be diminished, e.g., in designing new materials.
How does energy from an external source, such as impact or shock, initiate detonation? In general, it is believed to involve the formation of “hot spots” [3, 7, 15–20]. These are small regions in the crystal lattice in which is localized some portion of the externally–introduced energy; they are often associated with lattice defects: vacancies, voids, misalignments, dislocations, cracks, etc. If the hot spot energy is sufficient, it can initiate endothermal molecular processes that lead to self-sustaining exothermal chemical decomposition of the explosive [19–22]. Energy and gaseous products are released, and a high pressure shock wave may be created that propagates through the material at a supersonic velocity (detonation).
What are some of the endothermal molecular processes that can result in exothermal decomposition? Frequently invoked is the concept of a “trigger linkage” [13, 15], which is that the rupture of certain types of bonds is particularly likely to initiate detonation. Such bonds include C-NO2 in nitroaromatics, nitroaliphatics, and nitroheterocycles, O-NO2 in nitrate esters, N-NO2 in nitramines, and N-N2 in organic azides [3, 4, 15, 23].
Breaking a trigger linkage certainly needs to be considered as a possible endothermal initiating step. As discussed recently in detail [6], it is the basis – directly or indirectly – for many of the correlations that have been reported between sensitivity and assorted molecular properties, usually for compounds in a particular chemical category, e.g., nitroaromatics. (Overviews of such correlations have been given on several occasions [3, 5, 6, 12, 24].) However there are also other endothermal processes that may lead to exothermal decomposition. For example, various molecular rearrangements can occur [3–5, 13–15, 23, 25], including nitro/aci tautomerization, nitro/nitrite isomerization, furazan/furoxan formation, and others.
Accordingly, in addition to focusing upon specific bonds within molecules, there have also been extensive analyses of sensitivity in relation to a global feature of a molecule: the electrostatic potential produced by its nuclei and electrons. This is a physical observable, which can be determined experimentally by diffraction methods [26, 27] as well as computationally. The electrostatic potential is a fundamental determinant of molecular properties and behavior [28–31], and the potential computed on the molecular surface has been shown to be relevant to sensitivity. The surface is taken to be the 0.001 au (electrons/bohr3) contour of the molecule’s electronic density [32]. It was found that molecules of C,H,N,O-containing explosive compounds typically have an anomalous imbalance of positive and negative potentials on their surfaces: the central portions tend to be very positive, with weakly negative peripheries. This differs from most other organic molecules, in which the negative regions are often stronger although perhaps less extensive than the positive. This characteristic imbalance has been linked to sensitivity [3, 5, 6, 12, 33, 34]; for compounds of a given chemical type, sensitivity usually increases as the central portion becomes more positive.
There can be several reasons for such a link. In some cases, the anomalous potential imbalance may accompany the weakening of a trigger linkage [34], or another important bond. It may electrostatically promote key molecular rearrangements, or contribute to the formation of hot spots when the crystal lattice is deformed by an external force such as impact [6].
Any general discussion of sensitivity must take account of crystal lattice factors as well as molecular and physical ones. Quoting Doherty and Watt, “For more than two decades the question of the relationship between crystal properties and shock sensitivity in energetic materials has been a staple of energetics research programs around the world” [9]. The same can be said of impact sensitivity. The significant roles of lattice defects in hot spot formation have already been mentioned. Also to be noted is that polymorphs of a solid explosive can differ markedly in sensitivity [35, 36]. Our focus in the remainder of this paper will be upon some crystal lattice effects.
Sensitivity and free space in the crystal lattice: theory and experiment
The packing coefficient of a molecular solid is the fraction of the unit cell that is occupied by the molecules [37, 38]. It is given by,
in which Z is the number of molecules in the unit cell, Vint is the intrinsic gas phase molecular volume and Vcell is the cell volume. Vcell can readily be obtained from the molecular mass M, the crystal density ρ and Z:
Then, by combining Eqs. (1) and (2),
Vint presents more of a problem, because there is no rigorous definition of molecular volume. Since it is nevertheless a very useful concept, a number of procedures for assigning molecular volumes have been proposed [32, 37–40]. Eckhardt and Gavezzotti have concluded that packing coefficients for organic molecules in general are primarily between 0.6 and 0.8, averaging about 0.70 [41]. However for 43 C,H,N,O-containing energetic compounds, they found the packing coefficients to be higher; they ranged from about 0.71 to 0.83, and averaged 0.77. Eckhardt and Gavezzotti attributed this to the energetic molecules having fewer hydrogens.
These data suggest that roughly 15 %–30 % of the unit cell volumes of energetic compounds is free space. There is reason to believe, as shall be discussed, that this free space is among the factors that affect detonation initiation and hence sensitivity.
When an explosive compound is subjected to an external force, such as impact or shock, it undergoes rapid compression [16, 19, 42–46]. This causes an increase in temperature, which can be quantified by means of the Mie-Grüneisen equation [19, 46, 47]: the larger is the decrease in volume, the greater is the rise in temperature. Since the compressibility of a solid is anisotropic, the temperature increase will depend upon the direction in which the external force was exerted.
This newly-introduced thermal energy can lead to the formation of hot spots [16, 19, 42, 46, 48], which may result in the initiation of detonation. As mentioned earlier, hot spot formation is promoted by lattice defects, which also increases compressibility [44]. Rice et al. have proposed that a critical level of compression is required for detonation [43].
The preceeding considerations suggest a link between compressibility and sensitivity, and there is evidence to support this. Particularly notable are studies relating to the explosive pentaerythritol tetranitrate (PETN): Dick et al. [49–51] observed that it is much more sensitive to shock parallel to the [110] and [001] crystallographic directions than parallel to the [101] and [100]; Kunz later showed that PETN is more compressible in the [001] direction than in the [100] [52]. Piermarini et al. found the sensitivity of nitromethane to depend upon the orientation of the crystal relative to the applied stress [53]. It is also relevant that the very insensitive explosive TATB (1,3,5-triamino-2,4,6-trinitrobenzene) has a very low compressibility [54].
One of the factors governing compressibility is presumably the amount of free space in the crystal lattice. For example, we have estimated the free space in the crystal lattice of RDX (1,3,5-trinitro-1,3,5-triazacyclohexane) to be approximately 22 % of the volume [55]. In a computational investigation of the effect of pressure upon defect-free RDX, treated as being composed of rigid molecules, Kuklja and Kunz found that a relatively small initial pressure increase (isotropic) did cause the unit cell volume to rapidly diminish by about 20 % [44]. However reducing it further quickly became much more difficult; to decrease the volume by another 20 %, for instance, required roughly a 30-fold increase in the pressure. Thus, the region of “easy” compression closely matches the estimated free space.
In addition to its effect upon compressibility, the free space in the crystal lattice may also facilitate detonation initiation in another manner. Several computational studies indicate that the energies required to break the N-NO2 bonds in RDX [56] and HMX (1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane) [57, 58] and the C-NO2 in nitromethane [59, 60] are less when the molecules are at the crystal surfaces or by lattice voids than when they are in the bulk crystals. If such a bond can serve as a trigger linkage in a particular case, then having some neighboring free space would promote its rupture and the initiation of detonation.
Sensitivity and free space in the crystal lattice: a link
The preceding section presented a theoretical and experimental foundation for a link between sensitivity and compressibility/free space. Such a link had been proposed earlier [55, 61], but on an empirical basis. The total free space in the unit cell, denoted S, can be expressed in terms of the packing coefficient (the fraction of the unit cell that is occupied) by,
Inserting Eqs. (1) and (2) leads to,
The quantity M/ρ can be interpreted as the “effective” volume per molecule, Veff, by which is meant the hypothetical molecular volume that would correspond to the unit cell being completely filled, i.e., packing coefficient equal to one.
Thus,
The free space per molecule, labeled ΔV, is therefore,
Two questions now need to be addressed: (1) How should the intrinsic molecular volume Vint be determined? (2) Should a link be sought between sensitivity and the total free space S in the unit cell or the free space per molecule ΔV?
With regard to Vint, we follow the suggestion of Bader et al. [32] that molecular volume be taken to be the space encompassed by an appropriate outer contour of the molecule’s electronic density. This reflects the factors that are specific to the molecule, e.g., lone pairs, π electrons, strained bonds, etc. For instance, the 0.001 au (electrons/bohr3) contour has proven to be very effective for analyzing noncovalent interactions [30, 62–64]. The volume within this contour can also be utilized to obtain a rough approximation to the crystal density [65, 66], ρ ≈ M/V(0.001). This means, however, that V(0.001) is quite similar to Veff, Eq. (6), and therefore cannot be used to estimate the free space in the unit cell.
As our criterion for defining Vint we invoked Eckhardt and Gavezzotti’s conclusion that the packing coefficients of energetic compounds tend to be between 0.71 and 0.83, and to average 0.77 [41]. In our previous study [55], we tested several different electronic density contours to determine which gives volumes that, using Eq. (3), would best reproduce Eckhardt and Gavezzotti’s range of packing coefficients, and found that the 0.003 au contour does so very well. For a group of 21 explosive compounds, using Vint = V(0.003) in Eq. (3), we obtained values of 0.73–0.82 and an average of 0.77. Accordingly we take V(0.003) of the isolated molecule to be Vint. The physical significance of V(0.003) will be further discussed later in this section.
Proceeding to the second question, i.e., total free space S in the unit cell vs. free space per molecule ΔV, in our earlier work we used ΔV. This seemed reasonable because the endothermal processes that are believed to be likely possibilities for initiating detonation are unimolecular, e.g., trigger linkage rupture, molecular rearrangement, etc. This suggests a focus upon free space per molecule. In the present study we did revisit this issue, and investigated the use of S for a group of compounds but the results do not favor S over ΔV.
Our database, which is somewhat larger than those used previously [55, 61], is in Table 1. It lists, for 25 explosives, the experimentally-determined impact sensitivities and crystal densities. Impact sensitivities are commonly measured by dropping a mass m, often 2.5 kg, on a prepared sample of the compound [1, 2, 8, 9, 12]. The height from which 50 % of the drops result in evidence of reaction is designated as h50 and is indicative of the compound’s sensitivity. Since h50 depends upon the mass m, an equivalent approach that takes this into account is to give the impact energy, mgh50, where g is the acceleration due to gravity. For m = 2.5 kg and h50 = 100 cm, the impact energy is 24.52 J. The greater is h50, or the impact energy, the less sensitive to impact is the compound.
Table 1 also includes Veff, V(0.003) and ΔV for each explosive. The V(0.003) were computed at the density functional B3PW91/6-31G(d,p) level with Gaussian 09 [72] and the Wave Function Analysis–Surface Analysis Suite [73]. Veff and ΔV were evaluated with Eqs. (6) and (7).
What is the physical significance of V(0.003), which we are taking to be the intrinsic molecular volume Vint? To gain some insight into this, we examined the distances from atomic nuclei to the 0.003 au molecular electronic density contours in their immediate neighborhoods. This was done for 11 of the compounds in Table 1, and we also took carbon values from a recent study of model graphene systems [74]. These distances are compared in Table 2 with the van der Waals radii of the atoms. There is a distinct similarity, the largest differences being for hydrogen.
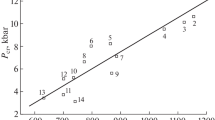
In Fig. 1, the measured impact sensitivities h50 of the 25 compounds in Table 1 are plotted against their computed ΔV. Overall, there is a general tendency for compounds having more free space per molecule in the crystal lattice (i.e., larger ΔV) to have greater sensitivity (i.e., lower h50). Figure 1 certainly does not exhibit a correlation, but it does support the concept that compressibility/free space is a factor – not always the dominant one – in determining sensitivity.

Measured impact sensitivity, h50, plotted against estimated free space per molecule in the unit cell, ΔV, for the 25 compounds in Table 1. Green squares: nitramines. Blue triangles: nitroaromatics. Red circles: non-nitramines and non-nitroaromatics
When Fig. 1 is examined in detail, the points are seen to fall into three structurally distinct subgroups:
-
(1)
For the eight nitramines, indicated by green squares, h50 decreases almost linearly but very weakly as ΔV increases [55, 61]. Most of the compounds are quite sensitive, with h50 < 35 cm, despite ΔV varying widely from 29 to 86 Å3; the highest h50 is only 55 cm. Thus the nitramine sensitivities depend to only a minor extent upon ΔV. It should be noted that we have included Tetryl and TNAZ with the nitramines even though they have C-NO2 bonds as well as N-NO2; this is in accordance with Kamlet and Adolph’s conclusion that impact behavior is governed by the most sensitive feature [14], and nitramines are typically among the more sensitive compounds [2]. A plot of h50 vs. ΔV for just the nitramines is in Fig. 2.
Fig. 2 
Measured impact sensitivity, h50, plotted against estimated free space per molecule in the unit cell, ΔV, for the eight nitramines in Table 1
-
(2)
The seven nitroaromatics, represented by blue triangles, are all within a relatively small range of intermediate ΔV values, between 48 and 61 Å3, whereas h50 covers a span of 47–141 cm. This is shown for the nitroaromatics alone in Fig. 3.
Fig. 3 
Measured impact sensitivity, h50, plotted against estimated free space per molecule in the unit cell, ΔV, for the seven nitroaromatics in Table 1
-
(3)
The remaining ten compounds, which are of diverse types, show a reasonably good relationship between h50 and ΔV (red circles), displayed separately in Fig. 4. The primary outlier is FOX-7.
Fig. 4 
Measured impact sensitivity, h50, plotted against estimated free space per molecule in the unit cell, ΔV, for the ten non-nitramines and non-nitroaromatics in Table 1



Discussion and summary
While Fig. 1 supports, overall, the concept of a link between sensitivity and free space per molecule, it also indicates that this link should be considered separately for at least three subgroups of compounds. Figures 2, 3, and 4 emphasize this conclusion.
The eight nitramines in Table 1 show a near-linear dependence of h50 upon ΔV (Fig. 2) but it is weak, reflecting the fact that these h50 do not differ greatly. Most of these compounds are quite sensitive and have relatively low h50; seven of the eight are below 35 cm. A high level of sensitivity is a feature of nitramines in general; 80 % of those listed by Storm et al. in their extensive compilation have h50 < 40 cm [2]. This may be due in part to the comparative weakness of N-NO2 bonds [5, 34, 77, 78], but there may also be other reasons. For example, Kamlet has drawn attention to autocatalysis as a factor in the sensitivities of aliphatic nitramines [13].
The h50 – ΔV pattern of the seven nitroaromatics in Table 1 is opposite to that of the nitramines. Figure 3 shows that the nitroaromatic h50 span nearly 100 cm despite the ΔV all being within an interval of just 13 Å3. Perhaps a ΔV of 50–60 Å3 is necessary but not sufficient for these compounds to undergo detonation initiation, and other factors determine what is sufficient in each case.
This might help to explain the well-known insensitivity of TATB, which has an h50 higher than the normally measurable limit [2]. Its ΔV is 38 Å3, which is well below the speculative necessary range.
In Fig. 4, FOX-7 is an outlier. This is perhaps not surprising, since FOX-7 differs in a key respect from most other explosives. The electrostatic potential on its molecular surface does not feature the characteristic strongly positive central region and weakly negative periphery that have been found to be linked to sensitivity, as already discussed. Instead there is a gradient in the surface electrostatic potential, from positive at the diamino end to negative at the dinitro [12]. This can be understood in terms of “push-pull” electronic delocalization in the molecule [79], for example:

Consistent with such delocalization is the fact that the C-C bond length, computed with the B3PW91/6-31G(d,p) procedure, is much longer in FOX-7 (1.426 Å) than in ethylene (1.329 Å). The atypical surface potential of FOX-7 may be related to its sensitivity being greater than its ΔV and Fig. 4 would suggest.
We want to emphasize, however, that we are not proposing Figs. 2 and 4 as correlations between h50 and ΔV. Our primary purpose in this work has been to demonstrate that the free space per molecule in the crystal lattice is one of the various factors that govern sensitivity, the significance of its role differing from one case to another. Figures 2 and 4 may be used cautiously to make tentative rough estimates of impact sensitivities for the respective categories of compounds, but this should be done with the understanding that ΔV may not be the determining factor in any given instance.
References
Iyer S, Slagg N (1988) In: Liebman JF, Greenberg A (eds) Structure and reactivity, ch 7. VCH, New York, pp 255–285
Storm CB, Stine JR, Kramer JF (1990) In: Bulusu SN (ed) Chemistry and physics of energetic materials, ch 27. Kluwer, Dordrecht, The Netherlands, pp 605–639
Politzer P, Murray JS (2003) In: Politzer P, Murray JS (eds) Energetic materials. Part 2. Detonation, combustion, ch 1. Elsevier, Amsterdam, pp 5–23
Zeman S (2007) Struct Bond 125:195–271
Politzer P, Murray JS (2014) In: Brinck T (ed) Green energetic materials. Wiley, Chichester, UK, doi: 10.1002/9781118676448
Politzer P, Murray JS (2014) Adv Quantum Chem 69:1–30
Armstrong RW, Coffey CS, DeVost VF, Elban WL (1990) J Appl Phys 68:979–984
Sučeska M (1995) Test methods for explosives. Springer, New York
Doherty RM, Watt DS (2008) Propell Explos Pyrotech 33:4–13
Elbeih A, Husarova A, Zeman S (2011) Cent Eur J Energ Mater 8:173–182
Wang Y, Jiang W, Song X, Deng G, Li F (2013) Cent Eur J Energ Mater 10:277–287
Rice BM, Hare JJ (2002) J Phys Chem A 106:1770–1783
Kamlet MJ (1976) Proceedings of the 6th Symposium (International) on Detonation, San Diego, CA, Report No, ACR 221, Office of Naval Research, Arlington, VA, pp 312–322
Kamlet MJ, Adolph HG (1979) Propell Explos 4:30–34
Kamlet MJ, Adolph HG (1981) Proceedings of the Seventh Symposium (International) on Detonation, Report No. NSWCMP-82-334, Naval Surface Warfare Center, Silver Springs, MD, pp 60–67
Tsai DH, Armstrong RW (1994) J Phys Chem 98:10997–11000
Tarver CM, Chidester SK, Nichols AL III (1996) J Phys Chem 100:5794–5799
White CT, Barrett JJC, Mintmire JW, Elert ML, Robertson DH (1996) Mat Res Soc Symp Proc 418:277–282
Dlott DD (2003) In: Politzer P, Murray JS (eds) Energetic materials. Part 2. Detonation, combustion. Elsevier, Amsterdam, pp 125–191, ch. 6
Shackelford SA (2008) Cent Eur J Energ Mater 5:75–101
Mader CL (1998) Numerical modeling of explosives and propellants, 2nd edn. CRC, Boca Raton, FL
Meyer R, Köhler J, Homburg A (2007) Explosives, 6th edn. Wiley-VCH, Weinheim, Germany
Brill TB, James KJ (1993) Chem Rev 93:2667–2692
Anders G, Borges I Jr (2011) J Phys Chem A 115:9055–9068
Zeman S (2003) In: Politzer P, Murray JS (eds) Energetic materials. Part 2. Detonation, combustion. Elsevier, Amsterdam, pp 25–52, ch 2
Stewart RF (1979) Chem Phys Lett 65:335–342
Klein CL, Stevens ED (1988) In: Liebman JF, Greenberg A (eds) Structure and reactivity. VCH, New York, pp 25–64, ch 2
Politzer P, Murray JS (2002) Theor Chem Acc 108:134–142
Ayers PW (2007) Chem Phys Lett 438:148–152
Murray JS, Politzer P (2011) WIREs Comp Mol Sci 1:153–163
Politzer P, Clark T (2005) Mol Phys 103:891–895
Bader RFW, Carroll MT, Cheeseman JR, Chang C (1987) J Am Chem Soc 109:7968–7979
Murray JS, Lane P, Politzer P (1998) Mol Phys 93:187–194
Murray JS, Concha MC, Politzer P (2009) Mol Phys 107:89–97
Kohno Y, Maekawa K, Tsuchioka T, Hashizume T, Imamura A (1994) Combust Flame 96:343–350
McCrone WC (1999) Chem&Eng News, July 5, p 2
Kitaigorodski AI (1961) Organic chemical crystallography. Consultants Bureau, New York
Dunitz JD, Filippini G, Gavezzotti A (2000) Tetrahedron 56:6595–6601
Stine JR (1990) J Energ Mater 8:41–73
Ammon HL (2001) Struct Chem 12:205–212, and references cited
Eckhardt CJ, Gavezzotti A (2007) J Phys Chem B 111:3430–3437
Tsai DH (1996) Mat Res Soc Symp Proc 418:281–286
Rice BM, Mattson W, Trevino SF (1998) Phys Rev E 57:5106–5111
Kuklja MM, Kunz AB (1999) J Appl Phys 86:4428–4434
Manaa MR (2003) In: Politzer P, Murray JS (eds) Energetic materials, Part 2. Detonation, combustion, ch 4. Elsevier, Amsterdam
Tarver CM, Urtiew PA, Tran TD (2005) J Energ Mater 23:183–203
Lemons DS, Lund CM (1999) Am J Phys 67:1105–1108
Maffre P, Peyrard M (1992) Phys Rev B 45:9551–9561
Dick JJ (1984) Appl Phys Lett 44:859–861
Dick JJ, Mulford RN, Spencer WJ, Pettit DR, Garcia E, Shaw DC (1991) J Appl Phys 70:3572–3587
Yoo CS, Holmes NC, Souers PC, Wu CJ, Ree FH, Dick JJ (2000) J Appl Phys 88:70–75
Kunz AB (1996) Mat Res Soc Symp Proc 418:287–292
Piermarini GJ, Block S, Miller PJ (1989) J Phys Chem 93:457–462
Zhang C (2007) J Phys Chem B 111:14295–14298
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2011) J Mol Model 17:2569–2574
Kuklja MM (2001) J Phys Chem B 105:10159–10162
Sharia O, Kuklja MM (2012) J Am Chem Soc 134:11815–11820
Sharia O, Kuklja MM (2012) J Phys Chem C 116:11077–11081
Roszak S, Keegstra PB, O’Neal DW, Hariharan PC, Kaufman JJ (1989) Int J Quantum Chem 36:353–368
Zhang C (2013) J Mol Model 19:477–483
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2010) J Mol Model 16:895–901
Politzer P, Murray JS (1998) J Mol Struct (Theochem) 425:107–114
Clark T, Murray JS, Lane P, Politzer P (2008) J Mol Model 14:689–697
Politzer P, Murray JS, Clark T (2013) Phys Chem Chem Phys 15:11178–11189
Politzer P, Martínez J, Murray JS, Concha MC, Toro-Labbé A (2009) Mol Phys 107:2095–2101
Rice BM, Byrd EFC (2013) J Comput Chem 34:2146–2151
Simpson RL, Urtiew PA, Ornellas DL, Moody GL, Scribner KJ, Hoffman DM (1997) Propell Explos Pyrotech 22:249–255
Archibald TG, Gilardi R, Baum K, George C (1990) J Org Chem 55:2920–2924
Wilson WS, Bliss DE, Christian SL, Knight DJ (1990) Explosive Properties of Polynitroaromatics, Report No NWC TP 7073, Naval Weapons Center, China Lake, CA
Gilardi RD, Butcher RJ (2001) Acta Cryst E57:657–658
Pagoria PF, Lee GS, Mitchell AR, Schmidt RD (2002) Thermochim Acta 384:187–204
Gaussian 09, Revision A.1, Frisch MJ.; Trucks GW, Schlegel HB, Scuseria GE, Robb MA et al. (2009) Gaussian Inc, Wallingford, CT
Bulat FA, Toro-Labbé A, Brinck T, Murray JS, Politzer P (2010) J Mol Model 16:1679–1693
Murray JS, Shields ZP-I, Lane P, Macaveiu L, Bulat FA (2013) J Mol Model 19:2825–2833
Rowland RS, Taylor R (1996) J Phys Chem 100:7384–7391
Bondi A (1964) J Phys Chem 68:441–451
Fried LE, Manaa MR, Pagoria PF, Simpson RL (2001) Annu Rev Mater Res 31:291–321
Mathieu D (2013) J Phys Chem A 117:2253–2259
Politzer P, Concha MC, Grice ME, Murray JS, Lane P (1998) J Mol Struct (Theochem) 452:75–83
Acknowledgments
We are pleased to join in honoring Tim Clark, a stimulating collaborator (e.g., refs. 31, 63, and 64) and a good friend. We greatly appreciate the support of this work by the Office of Naval Research, contract number N00014-12-1-0535, Program Officer Dr. Clifford D. Bedford.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to a Topical Collection on the occasion of Prof. Tim Clark’s 65th birthday
Rights and permissions
About this article

Cite this article
Politzer, P., Murray, J.S. Impact sensitivity and crystal lattice compressibility/free space. J Mol Model 20, 2223 (2014). https://doi.org/10.1007/s00894-014-2223-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2223-7