
Abstract
Although many 1,2,3,4-tetrazine-1,3-dioxide derivates have been synthesized, [1,2,5] oxadiazolo [3,4-e] [1,2,3,4]-tetrazine-4,6-di-N-dioxide (FTDO) is the only one with high enthalpy of formation and high detonation velocity. Whereas, its stability has not been studied. In the present work, the structure of FTDO was investigated using density functional theory (DFT) method, and its stability was calculated by potential energy surface scanning and structure interconvert thermodynamics under different temperatures. The spontaneous isomerization of FTDO and its effect on the stability of FTDO were investigated. The dissociation of FTDO to N2, N2O and furoxan fragments was studied, and the possibility of synthetic route from FTDO to TTTO was discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Since the first 1,2,3,4-tetrazine-1,3-dioxide derivatives were introduced in 1990 [1], many of them have been reported theoretically and experimentally [2–10], and the studies before 2004 have been summarized by Churakov [11]. However, only [1,2,5] oxadiazolo [3,4-e] [1,2,3,4]-tetrazine-4,6-di-N-dioxide (FTDO) could be called high energetic density compounds (HEDCs) among the synthesized compounds, though several other ones with higher predicted properties have been designed, such as TTTO, DTTO, etc., and their structures and predicted properties are listed in Table 1 [10, 12]. From then on N-oxides have also been arousing great interest because of the high detonation performance [14–16].
The first sample of FTDO was obtained by Aleksandr et al. [17] in 1995. Rezchikova et al. [18, 19] reported its IR and Raman spectra at the same year. Li et al. [20] designed a new synthetic route of FTDO with yield of 31 % in 2012, and its density and enthalpy of formation were measured in 2004 and 2011, with the value of 1.85 g cm−3 [5] and 160.9 kcal mol−1 [9] respectively. Security and combustion properties on this compound are studied. Teselkin et al. [8] reported the mechanical sensibility of FTDO in 2009. They found its mechanical sensibility was close to that of lead azide. In 2009 and 2011, Kalmykov et al. [21, 22] studied the combustibility of co-crystal of FTDO with 2,4-dinitro-2,4-diazapentane. They found it could combust stably at normal pressure, but unstably under several decades atmosphere pressure; and the higher the FTDO’s content is, the lower the critical pressure for stable combustion.
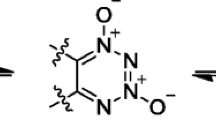
FTDO is not only a promising energetic molecule, but also an ideal intermediate for the synthesis of TTTO, one of the most desired high energetic density compounds. A procedure from FTDO to TTTO was proposed by Russian scientists about 10 years ago (see Scheme 1) [12]. However, there is no report on successful synthesis of TTTO up to now.

ᅟ
The stability of FTDO is essential for its applications in explosives or propellants. However, there are not systemic investigations on FTDO’s stability until now. The present work investigated the structure of FTDO further using density functional theory (DFT) method, and made systemic calculations on its stability by the method of potential energy surface scanning and structure interconverted thermodynamics under different temperatures. The synthesis feasibility of TTTO from FTDO was reconsidered.
Calculation details
DFT B3LYP method with cc-pvdz and 6-31G** basis set were carried out to investigate the structure of FTDO using Gaussian 09 program [23]. The geometries were fully optimized, and the frequency calculations were performed. The results indicated that these geometries correspond to be minima (no imaginary frequency) in the potential energy surfaces. FTDO’s dissociation barriers in different channels were calculated by the potential energy surface scanning for its dissociation routes and stability. The enthalpy change (ΔH), entropy change (ΔS), and Gibbs free energy change (ΔG) were calculated at different temperature (25-500 °C) to further confirm its stability.
Results and discussion
Structure
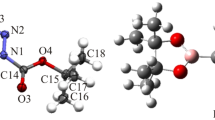
The optimized structure of FTDO with numbering atoms is presented in Fig. 1, and the bond lengths, bond angles, and dihedral angles are listed in Table 2.

Optimized geometric configuration of FTDO
It could be seen that the values of geometric parameters optimized at cc-pvdz and 6-31G** basis sets were nearly consistent. All atoms of FTDO molecule are approximatively in one plane, and the lengths of the bonds are between a typical single bond and a typical double bond to come into being a π conjugate system, which makes it aromatic.
Stability
In our present work, the stability of FTDO was investigated according to its dissociation, and the dissociation process was calculated and analyzed by the means of potential energy surface scanning (see Fig. 2) and Gibbs free energy changes (see Table 3). The results indicate FTDO would isomerize spontaneously (channel b) at ambient condition, in which the O(10) atom transfers from the tetrazine ring to the furazan ring, and the stability decreases after the isomerization. The calculated dissociation energy of the isomer (channel f) is only 13.27 kcal mol−1 (cc-pvdz) or 14.08 kcal mol−1 (6-31G**), with 12.95 kcal mol−1 (cc-pvdz) or 12.38 kcal mol−1 (6-31G**) lower than the dissociation energy of FTDO (channel c), which is 26.22 kcal mol−1 (cc-pvdz) or 26.46 kcal mol−1 (6-31G**). That means the isomer is much easier to dissociate than FTDO. According to the calculation results, the dissociation would begin at the N(7)-N(8) bond in tetrazine ring (channel f), and then be followed by the rupture of N(3)-O(5) bond in furazan ring (channel k). The dissociation energy of this process is 7.88 kcal mol−1 (cc-pvdz) or 6.62 kcal mol−1 (6-31G**). Ultimately, the C(2)-N(6) bond and C(1)-N(9) bond would break at the same time to generate N2, N2O, and furoxan fragments (channels n and o). This result is consistent with the mass spectrum data of FTDO [20] and the result of some preliminary research works carried out by other groups [24–27].

Potential energy scanning results (the data in parentheses is calculated at B3LYP/cc-pvdz basis set, and that in square brackets is calculated at B3LYP/6-31G**, unit: kcal mol−1)
The enthalpy change (ΔH), entropy change (ΔS), and Gibbs free energy change (ΔG) for all channels were calculated in the range of 25–500 °C to investigate the stability further (see Table 3). It could be concluded from the results that the data of Gibbs free energy change at room temperature for each channel is very nearly the energy calculated by potential energy surface scanning. The temperature has little effect on each channel, and the most difference of ΔG is only about 1 kcal mol−1. The conversion from FTDO to isomer 1 (channel b) is a spontaneous process, and the increasing temperature would make the process easier.
It could be seen from the above results that 1,2,3,4-tetrazine ring in FTDO would break first during its dissociation, which means the furazan ring in FTDO is more stable than the 1,2,3,4-tetrazine ring. As a result, the synthetic route from FTDO to TTTO designed by Russian scientist might be impracticable. The low energy barrier for the opening of 1,2,3,4-tetrazine in FTDO indicates its poor stability, and its application would be restricted under the specified conditions.
Conclusions
In the present work, the structure of FTDO was investigated using DFT method with cc-pvdz and 6-31G** basis sets. The results show that all atoms in FTDO are approximatively in one plane and the molecule is aromatic. The potential energy surface scanning and the thermodynamics calculation for FTDO were performed. The results indicated spontaneous conversion from FTDO to its isomer occurred and its stability was decreased, which would dissociate to N2, N2O, and furoxan fragments. So the stability of FTDO is poor, and its application would be restricted under specified conditions. The result also indicated the synthetic route from FTDO to TTTO might be infeasible because the furazan ring is more stable than the 1,2,3,4-tetrazine ring, and the tetrazine ring would open first during a reaction.
References
Churakov AM, Ioffe SL, Strelenko YA (1990) 1,2,3,4-tetrazine-1,3-dixoides-A new class of heterocyclic compounds. Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (39):639–640
Tartakovsky VA, Filatov IE, Churakov AM, Ioffe SL, Strelenko YA, Kuźmin VS, Rusinov GL, Pashkevich KI (2004) Synthesis and structures of pyridoannelated 1,2,3,4-tetrazine 1,3-dioxides. Russian Chemical Bulletin, International Edition 53(11):2577–2583
Smirnov OY, Churakov AM, Strelenko YA, Tartakovsky VA (2008) Benzo-1,2,3,4-tetrazine 1,3-dioxides annulated with tetraazapentalene systems 2. Annulation at the C(6)-C(7) bond. Russian Chemical Bulletin, International Edition 57(10):2180–2184
Song XL, Li JC, Hou H, Wang BS (2009) Extensive theoretical studies of a new energetic material: tetrazino-tetrazine-tetraoxide (TTTO). J Comput Chem 30:1816–1820
Lempert DB, Nechiporenko GN, Soglasnova SI (2004) Specific momentum of rocket propellants containing oxidizers based on C, N, and O atoms versus the enthalpy of formation and elementary composition of the oxidizer. Khim Fiz 23(5):75–81
Kiselev VG, Gritsan NP, Zarko VE, Kalmykov PI, Shandakov VA (2007) Multilevel quantum chemical calculation of the enthalpy of formation of [1,2,5] oxadiazolo [3, 4-e] [1, 2, 3, 4]-tetrazine-4, 6-Di-N-dioxide. Combustion, Explosion, and Shock Waves 43(5):562–566
Kalmykov PI, Burtsev YA, Kuznetsova NP, Konstantinov VV (2004) Phase state and features of formation of the structure of eutectic alloys based on DF-2. In: Proc. III All-Russian conf. Energetic Condensed Systems (Chernogolovka), Yaunus, Moscow, pp. 64–66
Teselkin VA (2009) Mechanical sensitivity of furazano-1,2,3,4-tetrazine-1,3-dioxide. Combustion, Explosion, and Shock Waves 45(5):632–633
Pepekin VI, Matyushin YN, Gubina TV (2011) Enthalpy of formation and explosive properties of 5,6-(3,4-furazano)-1,2,3,4-tetrazine-1,3-dioxide. Russian Journal of Physical Chemistry B 5(1):97–100
Politzer P, Lane P, Murray JS (2013) Computational characterization of two di-1,2,3,4-tetrazine tetraoxides, DTTO and iso-DTTO, as potential energetic compounds. Central European Journal of Energetic Materials 10(1):37–52
Churakov AM, Tartakovsky VA (2004) Progress in 1,2,3,4-tetrazine chemistry. Chem Rev 104:2601–2616
DR HAROLD SHECHTER. AFRL-SR-AR-TR-05-0032. 15 NOV, 2004.
Kamlet MJ, Jacobs SJ (1968) Chemistry of detonation. I. A simple method for calculating detonation properties of C,H,N,O explosives. The Journal of Chemical Physics 48:23–35
Politzer P, Lane P, Murray JS (2013) Tricyclic polyazine N-oxides as proposed energetic compounds. Central European Journal of Energetic Materials 10(3):305–323
Politzer P, Lane P, Murray JS (2013) Computational analysis of relative stabilities of polyazine N-oxides. Struct Chem 24:1965–1974
Politzer P, Lane P, Murray JS (2013) Some interesting aspects of N-oxides. Molecular Physics, http://dx.doi.org/10.1080/00268976.2013.854934.
Churakov AM, Ioffe SL, Tartakovsky VA (1995) Synthesis of [1,2,5] oxadiazolo [3,4-e] [1,2,3,4] tetrazine 4,6-di-N-oxide. Mendeleev, Communication, pp 227–228
Rezchikova KI, Churakov AM, Shlyapochnikov VA, Tartakovskii VA (1995) 1,2,3,4-tetrazine-1,3-di-N-oxides-novel high nitrogen compounds: vibrational spectra and structure. Mendeleev Communication 3:100–102
Rezchikova KI, Churakov AM, Shlyapochnikov VA, Tartakovskii VA (1995) Spectroscopic investigation of condensed 1,2,3,4-tetrazine-1,3-dioxides. Izv. Akad. Nauk, Ser. Khim., (11):2187–2189
Li XZ, Wang BZ, Li H, Li YN, Bi FQ, Huo H, Fan XZ (2012) Novel synthetic route and characterization of [1,2,5] oxadiazolo-[3,4-e][1,2,3,4] tetrazine 4,6-di-N-oxide (FTDO). Chinese Journal of Organic Chemistry 32(10):1975–1980
Zarko VE, Simonenko VN, Kalmykov PI (2009) Laser initiation of crystallized mixtures of furazanotetrazine dioxide and dinitrodiazapentane. Combustion, Explosion, and Shock Waves 45(6):752–755
Kalmykov PI, Zarko VE, Sidel’nikov AA (2011) Specific features of the crystal and phase structure of binary systems 5,6-(3’,4’-furazano)-1,2,3,4- tetrazine- 1,3- dioxide-2,4-dinitro- 2,4-diazapentane. Russ J Appl Chem 84(2):248–255
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian, Inc., Wallingford, CT
Pasinszki T, Westwood NPC (1996) Ground, excited, and ionic states of the NCCNO molecule: a hel photoelectron, infrared, ultraviolet, and ab initio investigation. The Journal of Physical Chemistry 100(42):16856–16863
Pasinszki T, Westwood NPC (2001) Gas-phase spectroscopy of the unstable acetonitrile N-oxide molecule, CH3CNO. The Journal of Physical Chemistry A 105(8):1244–1253
Havasi B, Pasinszki T, Westwood NPC (2005) Gas-phase infrared and ab initio study of the unstable CF3CNO molecule and its stable furoxan ring dimer. The Journal of Physical Chemistry A 109(17):3864–3874
Pasinszki T, Havasi B, Hajgató B, Westwood NPC (2009) Synthesis, spectroscopy and structure of the parent furoxan (HCNO)2. The Journal of Physical Chemistry A 113(1):170–176
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lai, WP., Lian, P., Yu, T. et al. Theoretical study on the structure and stability of [1,2,5] oxadiazolo [3,4-e] [1,2,3,4]-tetrazine-4,6-Di-N-dioxide (FTDO). J Mol Model 20, 2343 (2014). https://doi.org/10.1007/s00894-014-2343-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2343-0