
Abstract
Tert-butyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole-1-carboxylate is a significant intermediate of 1H-indazole derivatives. In this paper, the title compound is acquired through two substitution reactions. The structure is corroborated by FTIR, 1H and 13C NMR spectroscopy, and MS. In the meantime, the single crystal is detected by means of X-ray diffraction, calculated by exerting density functional theory (DFT), and subjected to the crystallographic and conformational analysis. The results of comparing the DFT calculated value with the X-ray diffraction value display that the optimized molecular structure does cohere with the single crystal structure ascertained via the experiment. The 98.28% stable conformer and 1.72% unstable conformers are found in the DFT calculations. Furthermore, to reveal the physicochemical features of the title compound, the molecular electrostatic potential and frontier molecular orbitals are investigated through DFT.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Nitrogen heterocyclic compounds widely exist in nature and organisms and attract great attention in the fields of medicine, pesticides, functional materials, and chemical engineering due to their unique biological activities, high-efficiency with low-toxicity, environmental friendliness, and structural diversity [1-3]. Thereinto, indazole derivatives have been reported to have anticancer, antiviral, antibacterial, antiprotozoal, antipsychotic, anti-inflammatory, analgesic, and radiosensitization effects [4-9]. Moreover, except for the areas of medicine, they also deserve further research in the fields of agriculture, energy, and others because of their insecticidal, weeding, and photoelectric activities [2, 10, 11]. As a significant intermediate of 1H-indazole substituted at the most extensively studied C3 and C5 positions, tert-butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole-1-carboxylate (1) can be aromatized at the C5 position through the Suzuki–Miyaura reaction, and the structure can be further modified to exploit the derivation of the indazole structure type [5, 12-14].
Based on the above viewpoints, we report the synthesis of compound 1 by two substitution reactions (Scheme 1).

Synthesis method of title compound 1.
The structure was characterized by means of FTIR, 1H and 13C NMR spectroscopy, and MS, and the single crystal was ascertained via X-ray diffraction (XRD). In the study, we calculated the optimal structure of compound 1 through the DFT method with the 6-311G(2d,p) basis set at the B3LYP level [15]. Afterwards, compound 1 was subjected to the conformational analysis, and its structure was detected by XRD and compared with the optimized structure. The comparison reveals that the optimized molecular structure does cohere with the crystal structure ascertained by single crystal XRD. Furthermore, the DFT method was employed to discuss the molecular electrostatic potential and frontier molecular orbitals of compound 1, demonstrating that compound 1 has good chemical stability and certain reactivity.
EXPERIMENTAL
General remarks
For the title compound, the IR spectrum was recorded on a Bruker IFS-55V IR spectrometer (Bruker, Germany), in the range of 4000-400 cm–1 with the KBr particle method and a 1.0 cm–1 resolution. 1H and 13C NMR spectra were recorded on a Bruker ARX-400, 400 MHz spectrometer (Bruker Bioscience, Billerica, MA, USA) with TMS as an internal control. Mass spectra were recorded in the ESI pattern on an Agilent 1100 LC-MS device (Agilent Technologies, Palo Alto, CA, USA). Further, the XRD data for the crystal were recorded on a Bruker APEX II X-diffractometer. Data collection: APEX2; cell processing: SAINT; the program employed to resolve the structure: SHELXL-2018/3; the program utilized to clarify the structure and describe molecular graphics: SHELXL-2018/3; the program employed to assess the centroid-centroid distance: Mercury 2.3. All the reagents and solvents were acquired from a commercial provider and utilized without further purification.
Synthetic procedure
Synthesis of tert-butyl 5-bromo-1H-indazole-1-carboxylate (3). In a 500 mL single-neck flask, 5-bromoindazole (2) (30.00 g, 152.26 mmol) was dissolved in acetonitrile (150 mL). Under stirring, di-tert-butyl dicarbonate (36.55 g, 167.47 mmol), 4-dimethylaminopyridine (3.72 g, 152.26 mmol), and triethylamine (30.00 g, 152.26 mmol) were added to the reaction mixture in turn and reacted at room temperature for 30 min (TLC monitoring). After the reaction was completed, the reaction mixture was decanted into 100 mL of a saturated aqueous citric acid solution and extracted with ethyl acetate (50 mL×3). The organic phases were combined and scrubbed with a saturated aqueous citric acid solution (40 mL×3). After drying with anhydrous sodium sulfate, the organic phases were concentrated under the reduced pressure to acquire compound 3 (40.51 g, yield 89.5%) as a yellow solid.
Synthesis of tert-butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole-1-carboxylate (1). In a 500 mL single-neck flask, compound 3 (22.00 g, 74.04 mmol) was dissolved in acetonitrile (150 mL). Under stirring, potassium acetate (19.62 g, 199.92 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (0.73 g, 1.00 mmol) were added. After the reaction mixture was stirred for 15 min at room temperature, bis(pinacolato)dibon (50.57 g, 199.14 mmol) was decanted to the reaction mixture. The reaction mixture was heated to 78 °C and circulation refluxed for 40 min by inches (TLC monitoring). After the reaction was completed, the reaction mixture was filtered with suction, and an appropriate amount of water was decanted to the filtrate, and then filtered with suction to acquire a brown solid. After the crude product was purified by heating with methanol, compound 1 (22.26 g, yield 87.3%) was ultimately obtained as some light brown solid. 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H, 13-CH), 8.23 (s, 1H, 10-CH), 8.11 (d, J = 8.4 Hz, 1H, 9-CH), 7.85 (d, J = 8.5 Hz, 1H, 7-CH), 1.65 (s, 9H, 16-CH, 17-CH, 18-CH), 1.32 (s, 12H, 2-CH, 3-CH, 4-CH, 6-CH); 13C NMR (101 MHz, DMSO-d6) δ 149.03(C=O), 141.20 (Ar–C), 140.73 (Ar–C), 134.85 (C=N), 129.40 (Ar–C), 126.02 (Ar–C), 124.18 (Ar–C), 114.02 (Ar–C), 85.10 (C–O), 84.35 (C–O), 28.17 (–CH3)), 25.20 (–CH3). MS (ESI): m/z = 367.18 [M+Na]+.
X-ray crystal structure determination. Compound 1 was dissolved in methanol, and the solvent was naturally evaporated tardily in air at room temperature. Organic matter precipitated slowly, and the single crystal of compound 1 suitable for the XRD analysis was acquired. The randomly selected light brown crystal with dimensions of 0.19×0.13×0.11 mm was installed on a glass fiber for data collection. The lattice constants and diffraction intensities of the title compound were measured with a Bruker APEX II diffractometer, the MoKα radiation source (λ = 0.71073 Å) monochromated by graphite at 170.0 K. Up to 22302 reflections were gathered in the range of 4.288 ≤ 2θ ≤ 52.77° (index ranges: –10 ≤ h ≤ 10, –23 ≤ k ≤ 23, –27 ≤ l ≤ 28) by a φ–ω scan mode and 3865 were independent with Rint = 0.0748, out of which 2393 were considered to be observed with I > 2σ(I) in the succeeding refinement [16]. The structure was solved by direct methods using SHELXT-2018/3 and refined on F2 through the full-matrix least-squares procedure for all data within SHELXL-2018/3 [17]. The hydrogen atoms were ascertained by theoretical calculations. The final refined result converges to the reliability factor R = 0.0492 and the weighted reliability factor wR = 0.1203 (\(w=1/[{{\sigma }^{2}}(F_{0}^{2})+{{(0.0306P)}^{2}}],\ \ \text{where }P=(F_{0}^{2}+2F_{\text{c}}^{2})/3,\) (Δ/σ)max = 0.000, S = 1.064 (Δρ)max = 0.158 e/Å3 and (Δρ)min = –0.175 e/Å3. The ORTEP illustration of the crystal structure of compound 1 is demonstrated in Fig. 1. The crystallographic and processed data are demonstrated in Table S1 (Supplementary Materials).
Crystallographic data of the title compound have been deposited with the Cambridge Crystallographic Data Centre (ССDC 2059118) and can be obtained gratis by accessing the website: www.ccdc.cam.ac.uk/data_request/cif.
Quantum chemistry/DFT calculation. In this study, all theoretical calculations of the title compound were performed with the Gaussian 09 software package in the ground state (vacuum), exerting the B3LYP method with the 6-311G(2d,p) basis set. Based on the optimized structures, geometrical, electronic, and energy parameters were excerpted from the GaussView 5.0 program [18-20].
RESULTS AND DISCUSSION
Synthesis and characterization. Compound 1 was acquired through two substitution reactions, as expected, and had a high yield (total yield: 78.1%). Its structure was corroborated by FTIR, 1H and 13C NMR spectroscopy and MS, as demonstrated in Supplementary Materials (Figs. S1–S4). It is worth noting that in the entire synthetic route from compound 3 to compound 1, post-processing is simple and does not require column chromatography.
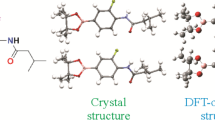
Crystallographic analysis. XRD provides basic data and sufficient structural information for the structure analysis of the grown crystal of compound 1. The title compound crystallizes in the orthorhombic space group Pbca with a = 8.7892(3) Å, b = 18.9939(7) Å, c = 22.6795(8) Å, α = β = γ = 90°, V = 3786.1(2) Å3, Z = 8, T = 170.0 K, μ(MoKα) = 0.084 mm–1, Dc = 1.208 g/cm3. The ORTEP illustration of the crystal structure of compound 1 is demonstrated in Fig. 1. The crystallographic and processed data are demonstrated in Supplementary Materials (Table S1).

Crystal and DFT-optimized structures of compound 1.

Crystal structure stacking (a) and hydrogen bonding (b) diagrams of compound 1.
As shown in Table S2 (Supplementary Materials), all bond lengths, bond and torsion angles obtained by the crystallographic analysis of the title compound match well with the calculation results with the DFT-optimized structure, and they are all within the normal range.
As shown in Fig. 2, the molecular packing of the crystal structure is formed by Van der Waals and hydrogen bonds. It should be noted that there is no π–π stacking interaction in the crystal structure. The centroid-centroid distances of Cg2…Cg2 and Cg2…Cg3 are 5.5820 Å and 5.5745 Å, in which Cg2 and Cg3 are centers of N1–N2–C13–C12–C11 and C17–C8–C10–C11–C12 rings, which are not within 4 Å [21]. In the crystal structure there are three intramolecular C10–H10…O(3) (2.457 Å), C16–H16A…O3 (2.433 Å), C17–H17C…O3 (2.421 Å) hydrogen bonds (Table 1) contributing to the crystal stability.
Conformational determination. Molecular conformation has a significant effect on the physical and chemical features of the molecule. Therefore, the calculable conformational analysis can help us better catch on the physical and chemical properties of organic molecules and elucidate some complex reaction phenomena. Through the Spartan 08 program, the initial conformational search for compound 1 was performed, exerting the molecular mechanics force field (MMFF) [16- 18]. Then, DFT/B3LYP/6-311G** was employed within the Gauss 09 package to perform the geometric optimization and frequency calculation for all possible conformations [15, 19]. According to the relative free energy, the percentage of each conformation was determined in the mixture equilibrated at room temperature (T = 295.15 K). Table 2 shows the Gibbs free energy (G), the relative Gibbs free energy (ΔG = exp(–Gi/RT), the gas constant R = 8.314 J/(mol·K), and the Boltzmann distribution (Boltzmann weighting factor \(Pi=\frac{\exp (-Gi/RT)}{\sum\nolimits_{j}{\exp }(-Gi/RT)}\times 100\%\)) of compound 1 in different conformers.
Fig. 3 shows the two conformational isomers of compound 1. The potential energy surface was scanned, and the summary of the potential surface revealed that compound 1 had two relatively stable conformers 1-1 (98.28%) and 1-2 (1.72%). The difference between the two conformations lies mainly in the tert-butyl carbonyl group. Since the N1–C14 bond is used as a rotating bond, the tert-butyl carbonyl group rotates about 180°. At the same time, the analysis suggests that conformation 1-1 occupies 98.18% because of the effect of intramolecular hydrogen bonds.
Molecular electrostatic potential (MEP). To better understand the intermolecular interaction region of compound 1, MEP of the main conformational molecules (with the same crystal structure) was studied by the B3LYP/6311G (2d,p) method [15]. In the MEP image, diverse colors show various electrostatic potentials on the molecular surfaces, and the increasing order of electrostatic potentials is blue > green > yellow > orange > red (see electronic version). The red color denotes the most potent repulsive force, and the blue color illustrates the most potent attractive force. As shown in Fig. 4, the positive regions are located around the H atoms of the methyl group of pinacol borate and tert-butyl carbonate and around the H atoms; these regions have a positive potential. Meanwhile, the negative potential regions are located at the electronegative atoms such as nitrogen and oxygen in association with their lone pairs.

Relatively stable and relatively unstable conformers of compound 1.

MEP diagram of compound 1.

HOMO and LUMO of compound 1.
Frontier molecular orbitals (FMOs). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are the chief molecular orbitals involved in the intramolecular charge transfer. The magnitude of the orbital energy gap between HOMO and LUMO reflects the ability of electrons to a transition from an occupied orbital to an empty orbital, and to a definite extent represents the aptitude of molecules to participate in chemical reactions [20]. In order to further study the chemical stability of the conformer (with the same crystal structure) with the B3LYP/6-311G(2d,p) method [15] along with the LUMO (ELUMO) and HOMO (EHOMO) energies and their orbital energy gap (ΔE). Fig. 5 shows FMOs and their positive and negative regions represented by red and green colors respectively. The ELUMO and EHOMO values are 1.3225 eV and –6.4790 eV, respectively, and the energy separation values between them are –5.1565 eV for consistency. For the calculated conformational isomers, a large LUMO–HOMO energy gap signifies a high excitation energy of the excited state, good chemical stability, and higher hardness. In addition, the ionization energy and the electron affinity can be expressed as: A = –ELUMO = 1.3225 eV, I = –EHOMO = 6.4790 eV. The hardness can be expressed as η = (I – A)/2, which represents the deformation of electron clouds in chemical systems with small perturbations encountered during the chemical process [22-24]. The obtained final hardness of compound 1 is 2.57852 eV.
CONCLUSIONS
In this paper, tert-butyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole-1-carboxylate was synthesized by two substitution reactions. Its structure was corroborated by the spectroscopic technique, NMR, MS, and single crystal XRD. The crystallographic studies and conformational analysis of the molecule of the title compound were carried out. According to the data displayed, the DFT calculated data correlate well with the XRD data obtained. In addition, the calculated data and experiments on MEP and FMOs show that the title compound has a certain chemical stability and reactivity, which play an important role in the pharmaceutical and industrial development of 1H-indazole derivatives.
REFERENCES
J. Akhtar, A.A. Khan, Z. Ali, R. Haider, and M.S. Yar. Eur. J. Med. Chem., 2017, 125, 143–189. https://doi.org/10.1016/j.ejmch.2016.09.023
P. P. Minieri and N. Y. Woodside. Patent US3637736, 1972.
N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso, and A. F. Trindade. Chem. Rev., 2009, 109, 2703–2802. https://doi.org/10.1021/cr800462w
T. Pemovska, E. Johnson, M. Kontro, G. A. Repasky, J. Chen, P. Wells, C. N. Cronin, M. McTigue, O. Kallioniemi,K. Porkka, B. W. Murray, and K. Wennerberg. Int. J. Oral Sci., 2015, 519, 102–105. https://doi.org/10.1038/nature14119
J. Y. Dong, Q. J. Zhang, Z. T. Wang, G. Huang, and S. S. Li. ChemMedChem, 2018, 13, 1490–1507. https://doi.org/10.1002/cdmc.201800253
I. Denya, S. F. Malan, and J. Joubert. Expert Opin. Ther. Pat., 2018, 28, 441–453. https://doi.org/10.1080/13543376.2018.1472240
H. Cerecetto, A. Gerpe, M. González, V. J. Arán, and C. O. de Ocáriz. Mini-Rev. Med. Chem., 2005, 5, 869–878. https://doi.org/10.2174/138955705774329564
J. M. Frost, D. A. DeGoey, L. Shi, R. J. Gum, M. M. Fricano, G. L. Lundgaard, O. F. El-Kouhen, G. C. Hsieh, T. Neelands, M. A. Matulenko, J. F. Daanen, M. Pai, N. Ghoreishi-Haack, C. C. Zhan, X. F. Zhang, and M. E. Kort.J. Med. Chem., 2016, 59, 3371–3391. https://doi.org/10.1021/acs.jmedchem.6b00063
R. X. Wang, P. J. Shi, B. Hu, E. C. Rao, F. S. Mi, Z. Li, and Y. Shen. Chin. Med. Sci. J., 1995, 1, 11. https://doi.org/CNKI:SUN:ZYKY.0.1995-01-002
Z. Ghamati, M. Pordel, A. Davoodnia, and S. A. Beyramabadi. Int. J. Energy Res., 2021, 45, 7797–7805. https://doi.org/10.1002/er.6363
O. S Kim, J. H. Jang, H. T. Kim, S. J. Han, G. C. Tsui, and J. M. Joo. Org. Lett., 2017, 19, 1450–1453. https://doi.org/10.1021/acs.orglett.7b00410
L. Rooney, A. Vidal, A. M. DSouza, N. Devereux, B. Masick, V. Boissel, R. West, V. Head, R. Stringer, J. Lao,J. P. Matt, P. Ardem, N. Mark, S. Natalie, P. Moh, V. J. Martin, M. S. Andrew, P. H. Michael, and C. T. David. J. Med. Chem., 2014, 51, 5129–5140. https://doi.org/10.1021/jm4019869
D. M. Fink, H. K. Smith, P. R. Eastwood, and H. Hunt. Patent WO086705, 2006.
P. W. Glunz, I. Delucca, and A. K. Dilger. Patent WO009625, 2018.
J. S. Zhao, P. Jin, N. Xi, and D. D. Wei. Chin. J. Struct. Chem., 2017, 36, 937–942. https://doi.org/10.14102/j.cnki.0254-5861.2011-1437
W. Y. Lin, F. Yang, A. N. Duan, W. W. You, and P. L. Zhao. Chin. J. Struct. Chem., 2018, 37, 1557–1562. https://doi.org/10.14102/j.cnki.0254-5861.2011-1967
G. M. Sheldrick. SHELXS-2014/7. University of Göttingen: Göttingen, Germany, 2014. https://doi.org/10.1007/S.10904-011-9556-p
G. Malecki. Polyhedron, 2010, 29, 2489–2497. https://doi.org/10.1016/j.poly.2010.05.019
M. Grabda, S. Oleszek-Kudlak, E. Shibata, and T. Nakamur. J. Mol. Struct: THEOCHEM, 2007, 822, 38–44. https://doi.org/10.1016/j.theochem.2007.07.013
A. Frish, A. B. Nielsen, A. J. Holder. Gauss View User Manual. Gaussian: Pittsburg, PA, 2011. https://doi.org/10.1021/ja1086485
S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikam, and K. Tanabe. J. Am. Chem. Soc., 2002, 124, 102–112. https://doi.org/10.1021/ja0105212
R. G. Parr and R. G. Pearson. J. Am. Chem. Soc., 1984, 15, 7512–7516. https://doi.org/10.1002/chin.198413001
R. G. Pearson. Proc. Natl. Acad. Sci., 1986, 83, 8440–8441. https://doi.org/10.1073/pnas.83.22.8440
R. G. Parr and P. K. Chattaraj. J. Am. Chem. Soc., 1991, 113, 1854–1855. https://doi.org/10.1021/ja00005a072
Funding
This work was supported by Guizhou Provincial Natural Science Foundation ([2020]1Y393) and the Science and Technology Program Platform for Talents of Guizhou province ([2018]5781).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 9, pp. 1454-1460.https://doi.org/10.26902/JSC_id79913
Rights and permissions
About this article

Cite this article
Ye, W.J., Chen, D.M., Wu, Q.M. et al. SYNTHESIS, CRYSTAL STRUCTURE, AND A DFT STUDY OF TERT-BUTYL-5-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-1H-INDAZOLE-1-CARBOXYLATE. J Struct Chem 62, 1357–1364 (2021). https://doi.org/10.1134/S0022476621090043
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621090043