
Abstract
The derivatives of 1,2,3,4-tetrazine may be promising candidates of high-energy density compounds and are receiving more and more attention. In this study, two 1,2,3,4-tetrazines, furoxano-1,2,3,4-tetrazine-1,3,5-trioxide (FTTO-α) and furoxano-1,2,3,4-tetrazine-1,3,7-trioxide (FTTO-β), were theoretically studied. The geometrical structures in gas phase were studied at the B3LYP/6-311++G(d,p) level of density functional theory (DFT). The gas phase enthalpies of formation were calculated by the homodesmotic reaction method. The enthalpies of sublimation and solid phase enthalpies of formation were predicted with corrections of electrostatic potential method at the B3PW91/6-31G(d,p) level. The detonation properties were estimated with the Kamlet-Jacobs equations based on the predicted densities and enthalpies of formation in solid state. The available free space in the lattice was calculated to evaluate their stability. Calculations of potential energy surface and structure interconversion thermodynamics under different temperatures were carried out to further confirm their stability. FTTOs have better performance than HMX and FTDO but are easy to decompose to 5,6-dinitroso-v-tetrazine 1,3-dioxide. A synthesis route for FTTO-β was proposed to provide a consideration for the further study. We believe FTTOs could be the key compounds to synthesize other v-tetrazines such as TTTO.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
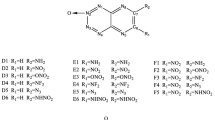
The energetic heterocyclic compounds, for example, furazan, tetrazole, and tetrazine derivatives have been widely used for military and civilian applications, and the synthesis and theoretical study of them have attracted considerable interest due to their high formation enthalpy and thermal stability [1–6]. As a typical nitrogen-rich heterocyclic compound, 1,2,3,4-tetrazine, also called vicinal-tetrazine or v-tetrazine, inherits all the advantages while it has its own speciality. Four directly-linkaged nitrogens bring more inner energy but also instability [7]. Based on the “alternating positive negative charge” (APNC) theory (Fig. 1), dozens of v-tetrazine 1,3-dioxide compounds with acceptable thermal stability have been successfully synthesized in the last 20 years [8, 9].

Stabilized v-tetrazine systems described by the APNC theory
5,6-(3,4-furazano)-1,2,3,4-tetrazine-1,3-dioxide (FTDO, 1), or [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide, was first described by Churakov et al. in 1995 [10]. The substance, with the density of 1.85 g cm−3 [11] and the enthalpy of formation of 160.9 kcal mol−1 [12], is of interest not only as a high-energy additive for increasing the specific momentum of rocket propellants but also as a component of energetic plasticizers of propellants [13]. Later, Zelenov and Lobanova et al. [14, 15] developed and studied new methods for the synthesis of FTDO from 4-(tert-butyl-NNO-azoxy)-N-nitro-1,2,5-oxadiazol-3-amine or its alkali metal salts and acid anhydrides (or chlorides) in the presence of strong acids, and discovered that the decomposition of FTDO in water proceeds with the formation of 5H-[1,2,3]triazolo[4,5-c][1,2,5]-oxadiazole and nitric acid (Scheme 1). Wang et al. [16] carried out a further study to the synthesis routine and obtained a series of high energy [1,2,3]triazolo[4,5-c][1,2,5]-oxadiazole salts.

ᅟ
FTDO is of great interest not only as a promising high energetic density compound (HEDC), but also an assumed intermediate for the synthesis of TTTO (2) [17], another promising v-tetrazine HEDC whose synthesis has not been reported yet. The route shown in Scheme 2 was proposed by Russian scientists about ten years ago [18]. However, different from most of the v-tetrazines, FTDO seems to have a more vulnerable v-tetrazine ring which makes it melt and decompose near 110 °C and be highly sensitive to mechanical impacts [19]. Disturbance of aromaticity in the molecule of FTDO resulted from the conjugation of the v-tetrazine ring with the furazan ring makes it possible for both nucleophilic and electrophilic species to attack the nitrogen atoms in the v-tetrazine ring [15], and the furazan ring would hardly rupture before the v-tetrazine ring under most of the conditions.

ᅟ
As one of the highly anticipated v-tetrazine HEDCs, furoxano-1,2,3,4-tetrazine-trioxide (FTTO), which structurally has one more coordination oxygen atom in furazan ring than FTDO, has been studied theoretically and experimentally few times since it was designed [17, 18, 20]. Compared to furazan, the furoxan brings higher enthalpy of formation and higher oxygen balance. By reduction with proper reducing agents, furoxan ring can be easily ruptured with the cleavage of the endocyclic N-O bond [21]. In the present study, two furoxano-1,2,3,4-tetrazine-trioxide isomers, furoxano-1,2,3,4-tetrazine-1,3,5-trioxide (FTTO-α) and furoxano-1,2,3,4-tetrazine-1,3,7-trioxide (FTTO-β) (Fig. 2), have been studied. Theoretical methods such as density functional theory (DFT) have been employed to predict their properties, such as molecular structures, enthalpies of formation, densities, detonation performance, and stability. A synthetic route was proposed. We believe FTTOs are key intermediates for further synthesis of other v-tetrazine HEDCs and our theoretical investigations on them will be helpful for further study.

Molecular structures of FTTO-α and FTTO-β
Computational methods
Geometry optimization of the molecular structures were carried out at the B3LYP/6-311++G(d,p) level with the Gaussian program package [22]. The vibrational analysis was performed at the same level. The optimized structures were confirmed to be the local minimum without imaginary frequencies. According to the previous study, the calculated frequencies were scaled uniformly by 0.96 to approximately correct the systematic overestimation.
The enthalpies of formation were calculated from the designed homodesmotic reactions (1) and (2) [23].


where ΔH°298 is the standard enthalpy change at 298 K; ΣΔf H°298,P, ΣΔf H°298,R, ΔΕ°298, and Δ(pV) represent the sum of enthalpies of formation of products, the sum of enthalpies of formation of reactants, the change in the total energy at 298 K, and the change in the product of pressure p and volume V, respectively.
where ΔΕ 0, ΔΕ ΖPV and ΔH°T denote the changes in the total energy at 0 K, in the zero-point vibrational energy, and in the temperature dependent enthalpy, respectively. Equation (5) assumes that substances in the reaction are all ideal gases.
Equations (6) and (7) were used to calculate the enthalpies of formation in solid state from the enthalpies of formation in gas phase and the sublimation enthalpies of FTTOs [24], the surface properties for prediction of sublimation enthalpies were computed at the B3PW91/6-31G(d,p) level of DFT.
where A S is the molecular surface area, ν = σ+ 2σ− 2/(σtot 2)2, σtot = σ+ 2 + σ− 2, σ+ 2 and σ− 2 are the mean variances of the positive and negative molecular electrostatic potential, respectively, α 1, α 2, and α 3 are the empirical parameters taken from [24].
The densities ρ (g cm−3) of FTTOs in solid state were computed by the improved Eq. (8) proposed by Politzer et al. at the B3PW91/6-31G(d,p) level [25], where M and V represent molecular weight (g mol−1) and volume of the 0.001 electrons bohr−3 contour of electronic density of the molecule (cm3 per molecule), respectively. The coefficients β 1, β 2, and β 3 are 0.9183, 0.0028, and 0.0043, respectively.
The empirical Kamlet-Jacobs equations [26–28] were used to estimate two important parameters of energetic materials, i.e., the detonation velocity (D) and detonation pressure (P).
where N is the moles of gaseous detonation products per gram of explosives, \( \overline{M} \) is the mean molecular mass of the detonation products, and Q is the detonation energy (cal g−1). N, \( \overline{M} \) and Q are determined based on the most exothermic principle. The products are supposed to be only CO2, H2O,and N2, so released energy in the decomposition reaction reaches its maximum.
To evaluate the stability of explosive molecules, some molecular structure related parameters, such as molecular electrostatic potential [29], Mulliken charge on nitro group [30], bond order [31], or bond dissociation energy of X-NO2 (X = C, N, O) [32], have been employed to estimate the shock sensitivity of nitro-containing explosives or propellants [33–35]. However, sensitivity depends upon a number of factors and it is not clear which one(s) may be dominant in any particular case [36]. There is evidence that one of the factors related to sensitivity is the compressibility of the solid compound [37–41], and that sensitivity increases with the amount of free space available in the crystal lattice [42]. According to Politzer’s suggestions [36, 42, 43], free space ΔV can be formulated as follows:
where V eff is the effective volume of the molecule that would correspond to 100 % packing of the unit cell, V int is the space encompassed by the 0.003 au contour of the molecule’s electronic density, M is the molecular mass and ρ is the crystal density.
Dissociation routes, dissociation energy barriers, and stability of FTTOs were predicted by scanning the pyrolysis potential energy surface. The enthalpy change (Δr H m) and Gibbs free energy change (Δr G m) under different temperatures (200-800 K) were calculated to further confirm their stability.
Results and discussion
Molecular structures and electrostatic potential surfaces
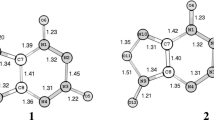
Geometric structure is the basic information that we can learn from a compound. The optimized molecular structure of FTDO, FTTO-α, and FTTO-β are shown in Fig. 3 for comparison.

Molecular geometrical parameters of FTDO, FTTO-α, and FTTO-β
All three molecules have planar structures. This planarity, as well as the endocyclic bond distances, indicates an extensive delocalization of the π electrons. The bond parameters of the tetrazine ring in FTTOs are close to those in FTDO. We can also notice the arresting length of 1.529 Å of the N5-O6 bond in FTTO-α. The value is far bigger than the normal length of N-O bond in furoxan, which probably means the furoxan ring in FTTO-α will be rather unstable and can easily decompose, or hardly exist. The O6-N7 bond length of 1.475 Å in FTTO-β is in the acceptable range. This may be from the electronegativity confliction between furoxan and 1,2,3,4-tetrazine 1,3-dioxide which was designed based on the APNC theory (Fig. 4). The positively charged N1 and N3, as well as the shortened N-O bond lengths, indicate the participation of the electrons on these oxygens in respective N-O bonds, which increases the stability of the compound by preventing the cleavage of the N-N single bond and the tetrazine ring. However, instead of N2-N3 bond (1.463 Å) in FTDO, O6-N7 (1.475 Å) is the longest bond in FTTO-β which may be the trigger bond.

APNC systems in FTTO-α and FTTO-β
The surface potentials of FTDO, FTTO-α, and FTTO-β are presented in Fig. 5. Positive values are strongly around the central portions of the molecules, as is characteristic of energetic compounds [44, 45], and the negatives around the peripheries. The negative surface potentials are due to the oxygens and the unsubstituted ring nitrogens. The surface potentials are positive around all nitrogens, and are more obvious for the ones bearing oxygens. The unsubstituted nitrogens have less regions of positive surface potentials than the substituted ones. It is fair to say that the unsubstituted nitrogens have more negative (or less positive) potentials than the substituted ones, which is consistent with the APNC systems in FTTO-α and FTTO-β.

Computed electrostatic potential on the 0.001 au molecular surface at B3LYP/6-311++G(d,p) level, with values −0.025 to +0.025 a.u.. Color coding: red, negative; yellow, slightly negative; green, neutral; light blue, slightly positive; dark blue, positive
Enthalpy of formation
The enthalpy of formation is the key property for energetic materials, by which we can estimate their energy output performance. Many methods have been used to predict the enthalpy of formation, such as semiempirical molecular orbital methods, atomization reaction method and isodesmic reaction method. Here the homodesmotic reactions (1) and (2), in which the chemical environments of the reactants and products are similar, were used to calculate the enthalpy of formation of FTTO-α and FTTO-β.
Table 1 lists the calculated total energies at 0 K (E 0) for five compounds in reactions (1) and (2), with some of their enthalpies of formation (Δf H°S). Additional calculations have been carried out using the G3 theory to get the accurate Δf H°S of furoxan and 1,2,3,4-tetrazine 1,3-dioxide through the atomization reaction CaHbOcNd(g) → aC(g) + bH(g) + cO(g) + dN(g), because their experimental Δf H°S are unavailable. The zero-point vibration energy (E ZPV), the temperature dependent thermal enthalpy change (ΔH°T), and the calculated enthalpies of formation in both gas and solid phases of two FTTO isomers are listed in Table 2.
Detonation parameters
The calculated detonation heats, velocities and pressures according to Eqs. (8), (9), and (10) are shown in Table 3. The detonation parameters of FTDO and typical explosives (HMX and CL-20) are also provided for comparison. We can find that FTTOs are more powerful than the commonly used explosive HMX and less powerful than CL-20. The detonation properties of FTTOs are also slightly better than those of FTDO.
Sensitivity
In the molecule of 1,2,3,4-tetrazine, the single N-N bond is the weakest chemical bond, which is usually considered as the key factor that determines the stability of 1,2,3,4-tetrazine compounds. According to our calculations, the bond dissociation activation energy (BDAE) of the central N-N single bond is about 250 to 280 kJ mol−1 while that of two N=N double bonds is around 290 kJ mol−1 for some 1,2,3,4-tetrazine compounds [49]. The BDAEs of the central N-N single bond of FTDO and FTTOs are 252.05 kJ mol−1 and 246.79 kJ mol−1, respectively. However, for FTTOs, the trigger linkage to explosion may be different because the O-N bond in the furoxan near the coordinated oxygen atom is the longest chemical bond and a rather fragile one.
Calculations of potential energy surface and structure interconversion thermodynamics under different temperatures were carried out to further confirm their stability. Figure 6 demonstrates that in the ring-opening reaction, the cleavage of the furoxan ring in FTTO-α needs only 45.8 kJ while FTTO-β must cross a transition state with an activation barrier of 60.1 kJ to become 5,6-dinitroso-v-tetrazine 1,3-dioxide. Both energies are far less than that needed for the cleavage of their v-tetrazine rings. Relatively, FTTO-β is more stable than FTTO-α. Figure 7 shows the thermodynamic parameters of two reactions from FTTO-α and FTTO-β to 5,6-dinitroso-v-tetrazine 1,3-dioxide at different temperatures (T = 200-800 K).

Gas-phase relative energies of FTTO-α and FTTO-β to 5,6-dinitroso-v-tetrazine 1,3-dioxide

Δr H m and Δr G m at different temperatures for ring-opening reactions of FTTO-α (left) and FTTO-β (right)
For both reactions, Δr G m decreases continuously while Δr H m increases slowly with the increasing temperature. The reactions are exothermic and Δr G ms are always negative in 200–800 K and are more negative at higher temperatures, therefore, high temperatures are more preferred for the two ring-opening reactions.
Sometimes, the BDAE or the bond dissociation energy (BDE) of ring-shaped or net-shaped molecules is hard to be accurately figured out [35] because of the presence of ring strains or conjugation. Politzer and co-workers [42] have shown that there is a rough relationship between the available free space in the lattice (ΔV) and the experimental impact sensitivity (H 50), and the sensitivity tends to increase as ΔV becomes large. This relationship has been applied to estimate the impact sensitivities of many promising energy compounds.
The ΔVs of FTDO, FTTO-α, FTTO-β, and several typical explosives are shown in Table 4 with the available experimental impact sensitivity for comparison. It is seen that FTTO-α and FTTO-β have ΔV value close to FTDO, which has the mechanical sensitivity close to lead azide [19]. According to the values of ΔV, FTTOs are less sensitive than HMX. However, FTTOs and HMX are two different species, the formers are heterocyclic compounds and the latter is a nitramine compound, and the free space gives only a rough estimate of the sensitivity, basically, the sensitivities of FTTOs, FTDO, and HMX are at the same level.
Possible synthetic route
V-tetrazine 1,3-dioxides are generally synthesized from aromatic amines with an ortho amino tert-butyl-NNO structure in which the tert-butyl group is the leaving group. Method of nitrating (e.g., with N2O5, NO2BF4), phosphorylating (e.g., with P4O10, PCl5), sulfonating (with SO3), and acylating (with AcCl/AlCl3, Ac2O/H2SO4 etc.) were successfully used to prepare v-tetrazine 1,3-dioxides of the aromatic series [14, 50–55]. Amino-(tert-butyl-NNO-azoxy) furoxan is unknown until last year [56], when Churakov and his colleagues experimentally confirmed the feasibility of the routine (Scheme 3). However, FTTOs were not mentioned in the work.

ᅟ
Although the yield of 4-aminofuroxan is only 1 % for the last step, we suggest using it as the starting material for the synthesis of FTTO-β because of the relatively higher stability of FTTO-β. The proposed synthesis route is shown in Scheme 4. In consideration of the possibility of isomerization or ring-opening, we believe it is better to deal with 4-aminofuroxan with softer cyclization reaction conditions such as HNO3-H2SO4-Ac2O system under 20 °C. We presume that 4-aminofuroxan (3) will be first nitrated to 4-nitroaminofuroxan (4), then attacked by ionic systems Ac2O-H2SO4 at the N-nitroamine fragment to yield the intermediate oxodiazonium ion (5) which undergoes a cyclization to FTTO-β via the elimination of tert-butyl (Scheme 5). Reaction (13) is the key step which determines whether the cyclization is practical. To estimate the thermodynamic possibility of the last step from (4) to the target product, the changes in thermodynamic functions of this reaction in the solvent of Ac2O at different temperatures (T = 200-800 K) were calculated. The results are presented in Fig. 8.

ᅟ

ᅟ

Δr H m and Δr G m at different temperatures for cyclization reaction of FTTO-β in Ac2O solvent
Δr G m decreases continuously and while Δr H m increases slowly with the increasing temperature. Δr G m is about −60 kJ mol−1 at 293.15 K. The reaction is endothermic at low temperature and exothermic above 250 K and will release a small amount of heat at room temperature. In conclusion, the proposed reaction is quite possible at ambient temperature. However, according to the changes in free energy we can only learn the thermodynamic possibility of the reactions, it is more practical to use activation energies to predict the feasibility of the reactions, this will be considered in our future synthetic and theoretical research works.
Conclusions
-
1.
With the help of DFT calculations, the properties of FTTOs were predicted. It was confirmed that FTTO-α and FTTO-β are interesting materials with the density >1.90 g cm−3, the detonation velocity >9000 m s−1 and detonation pressure >40 GPa. They show better performance than HMX and FTDO.
-
2.
FTTOs may be very sensitive. FTTO-α is less stable than FTTO-β. Although the BDAE of cleavage of v-tetrazine ring is about 246 kJ mol−1, the cleavage of furoxan ring needs only 45.8 kJ mol−1 for FTTO-α and 60.1 kJ mol−1 for FTTO-β. The results of ΔV also confirmed that FTTOs are less stable and have higher impact sensitivity than FTDO.
-
3.
According to the characteristics of FTTOs, we believe, FTTOs, if prepared successfully, could be the key material to synthesize monocyclic v-tetrazine dinitro-v-tetrazine 1,3-dioxide (DNTDO) and other v-tetrazine HEDCs such as TTTO (Scheme 6). We suggest choosing the relatively more stable FTTO-β as the research target and the thermodynamic possibility of the proposed synthesis route has been proved theoretically.
Scheme 6 
ᅟ

References
Tang Y, Yang H, Cheng G (2013) Synthesis and characterization of a stable, Catenated N11 energetic salt. Angew Chem Int Ed 52:1–4
Wu Q, Zhu W, Xiao H (2014) A new design strategy for high-energy Low-sensitivity explosives: combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule of 1-amino-5-Nitrotetrazole-3N-oxide. J Mater Chem A 2:13006–13015
Politzer P, Lane P, Murray JS (2013) Computational analysis of relative stabilities of polyazine N-oxides. Struct Chem 24:1965–1974
Liu Z, Wu Q, Zhu W (2013) Theoretical study of energetic trinitromethyl-substituted tetrazole and tetrazine derivatives. J Phys Org Chem 26:939–947
Jorgensen KR, Oyedepo GA, Wilson AK (2011) Highly energetic nitrogen species: reliable energetics via the correlation consistent composite approach (ccCA). J Hazard Mater 186:583–589
Jaidann M, Roy S, Abou-Rachid H (2010) A DFT theoretical study of heats of formation and detonation properties of nitrogen-rich explosives. J Hazard Mater 176:165–173
Thomas JR, Quelch GE, Schaefer HF III (1997) The unknown unsubstituted tetrazines:1,2,3,4-tetrazine and 1,2,3,5-tetrazine. J Org Chem 56:539–543
Churakov AM, Tartakovskii VA (2004) Progress in 1,2,3,4-tetrazine chemistry. Chem Rev 104:2601–2616
Bi F, Wang B, Li X, Fan X, Cheng X, Ge Z (2012) Progress in the energetic materials based on 1,2,3,4-tetrazine 1,3-dioxide. Chin J Energ Mater 5:630–637
Churakov AM, Ioffe SL, Tarakovsky VA (1995) Synthesis of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-Di-N-oxide. Mendeleev Commun 5:227–228
Lempert DB, Nechiporenko GN, Soglasnova SI (2004) Specific momentum of rocket propellants containing oxidizers based on C, N and O atoms versus the enthalpy of formation and elementary composition of the oxidizer. Khim Fiz 23:75–81
Pepekin VI, Matyushin YN, Gubina TV (2011) Enthalpy of formation and explosive properties of 5,6-(3,4-furazano)-1,2,3,4-tetrazine-1,3-dioxide. Russ J Phys Chem B 5:97–100
Kalmykov PI, Burtsev YA, Kuznetsova NP, Konstantinov VV (2004) Phase state and features of formation of the structure of eutectic alloys based on DF-2. Proc III All-russian conf energ condensed systems (Chernogolovka). Yaunus, Moscow, pp 64–66
Zelenov VP, Lobanova AA, Sysolyatin SV, Sevodina NV (2013) New syntheses of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide. Russ J Org Chem 49:467–477
Zelenov VP, Lobanova AA, Lyukshenko NI, Sysolyatin SV, Kalashnikov AI (2008) Behavior of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide in various media. Russ Chem Bull Int Ed 57:1384–1389
Li X, Wang B, Li Y, Li H, Zhou C, Zhang Y, Lian P (2013) Synthesis of 5H-[1,2,3]triazolo[4,5-c][1,2,5]oxdiazole and its energetic derivatives. Chin J Energ Mater 21:717–720
Lai W, Lian P, Yu T, Bu J, Liu Y, Zhu W, Lv J, Ge Z (2014) Theoretical study in the structure and stability of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]-tetrazine-4,6-Di-N-dioxide (FTDO). J Mol Model 20:2343
Shechter H, Venugopal M, Srinivasulu D (2006) Syntheses of 1,2,3,4-tetrazine Di-N-oxides, pentazole derivatives, pentazine poly-N-oxides, and nitroacetylenes. Ohio State University Research Foundations, DARPA/AFOSR Sponsored, Project 746566. March 8
Teselkin VA (2009) Mechanical sensitivity of furazano-1,2,3,4-tetrazine-1,3-dioxide. Combust Explo Shock 45:632–633
Politzer P, Lane P, Murray JS (2013) Some interesting aspects of N-oxides. Mol Phys http://dx.doi.org/10.1080/00268976.2013.854934
Sheremetev AB, Makhova NN (2001) Monocyclic Furazans and furoxans. Adv Heterocycl Chem 78:66–188
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, revision B.05. Gaussian, Wallingford
Wheeler SE, Houk KN, Schleyer PVR, Allen WD (2009) A hierarchy of homodesmotic reactions for thermochemistry. J Am Chem Soc 131:2547–2560
Politzer P, Ma Y, Lane P, Concha MC (2005) Computational prediction of standard Gas, liquid, and solid-phase heats of formation and heats of vaporization and sublimation. Int J Quantum Chem 105:341–347
Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbe A (2009) An electrostatic interaction correction for iimproved crystal density prediction. Mol Phys 107:2095–2101
Kamlet M, Jacobs SJ (1968) Chem Phys 48:23
Xiao HM, Xu XJ, Qiu L (2008) Theoretical design of high energy density materials. Science, Beijing
Politzer P, Murray JS (2011) Some perspective on estimating detonation properties of C, H, N, O compounds. Cent Eur J Energ Mater 8:209–220
Politzer P, Murray JS. Relationships between dissociation energies and electrostatic potentials of C-NO2 bonds: applications to impact sensitivities. J Mol Struct 376: 419–424
Zhang C, Shu Y, Huang Y, Zhao X, Dong H (2005) Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds. J Phys Chem B 109:8978–8982
Xiao H, Li Y (1995) Banding and electronic structures of metal azides—sensitivity and conductivity. Sci China B 38:538–545
Politzer P, Murray JS, Concha MC (1998) C-H and C-NO2 dissociation energies in some azines and Nitroazines. J Phys Chem A 102:6697–6701
Cao X, Xiang B, Zhang C (2012) Review on relationships between the molecular and crystal structure of explosives and their sensitivities. Chin J Energ Mater 5:643–649
Tan B, Long X, Peng R, Li H, Jin B, Chu S (2011) On the shock sensitivity of explosive compounds with small-scale Gap test. J Phys Chem A 115:10610–10616
Tan B, Long X, Li J, Nie F (2012) Insight into shock-induced chemical reaction from the perspective of ring strain and rotation of chemical bonds. J Mol Model 18:5127–5132
Politzer P, Lane P, Murray JS (2013) Tricyclic polyazine N-oxides as proposed energetic compounds. Cent Eur J Energ Mater 10:305–323
Dlott DD (2003) Fast molecular aspects in energetic materials. In: Politzer P, Murray JS (eds) Energetic materials. Part 2. Detonation, combustion, vol 6. Elsevier, Amsterdam, pp 125–191
Tsai DH, Armstrong RW (1994) Defect-enhanced structural relaxation mechanism for the evolution of Hot spots in rapidly compressed crystals. J Phys Chem 98:10997–11000
Kunz AB (1996) An Ab initio investigation of crystalline PETN. Mater Res Soc Symp Proc 418:287–292
Rice BM, Mattson W, Trevino SF (1998) Molecular-dynamics investigation of the desensitization of detonable material. Phys Rev E 57:5106–5111
Tarver CM, Urtiew PA, Tran TD (2005) Sensitivity of 2,6-diamino-3,5-dinitropyrazine-1-oxide. J Energ Mater 23:183–203
Pospisil M, Vavra P, Concha MC, Murray JS, Politzer P (2011) Sensitivity and the available free space per molecule in the unit cell. J Mol Model 17:2569–2574
Politzer P, Lane P, Murray JS (2013) Computational characterization of Two Di-1,2,3,4-tetrazine Tetraoxides, DTTO and iso-DTTO, as potential energetic compounds. Cent Eur J Energ Mater 10:37–52
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106:1770–1783
Murray JS, Lane P, Politzer P (1998) Effects of strongly electron-attracting components on molecular surface electrostatic potentials: applications to predicting impact sensitivities of energetic molecules. Mol Phys 93:187–194
Lide DR (2009) CRC Handbook of chemistry and physics 2010th edn. CRC, Boca Raton
He L, Dong L, Zhang G, Tan B, Huang M, Tao G (2012) Structure and properties of furazano[3,4-e]-1,2,3,4-tetrazine-1,3-dioxide. Chin J Energ Mater 6:693–696
Teipel U (2005) Energetic materials—particle processing and characterization. Wiley-VCH, Weinheim
Wang T, Zheng C, Yang J, Zhang X, Gong X, Xia M (2014) Theoretical studies on a new high energy density compound 6-amino-7-Nitropyrazino[2,3-e][1,2,3,4]tetrazine 1,3,5-trioxide (ANPTTO). J Mol Model 20:2261–2271
Voronin AA, Zelenov VP, Churakov AM, Strelenko YA, Fedyanin IV, Tartakovsky VA (2014) Synthesis of 1,2,3,4-tetrazine 1,3-dioxides annulated with 1,2,3-triazoles and 1,2,3-triazole 1-oxides. Tetrahedron 70:3018–3022
Churakov AM, Ioffe SL, Strelenko YA (1990) 1,2,3,4-tetrazine 1,3-dioxides—a new class of heterocyclic compounds. Rus Chem Bull 39:639–640
Churakov AM, Ioffe SL, Tartakovsky VA (1991) The first synthesis of 1,2,3,4-tetrazine −1,3-di-N-oxides. Mendeleev Commun 1:101–103
Frumkin AE, Churakov AM, Strelenko YA (2000) New approach to the synthesis of benzo[e][1,2,3,4]tetrazine 1,3-dioxides. Rus Chem Bull 49:482–486
Frumkin AE, Churakov AM, Strelenko YA (1999) Synthesis of 1,2,3,4-tetrazino[5,6-f]benzo-1,2,3,4-tetrazine 1,3,7,9-tetra-N-oxides. Org Lett 5:721–724
Tartakovsky VA, Churakov AM, Ioffe SL, Strelenko YA (2004) Synthesis and structures of pyridoannelated 1,2,3,4-tetrazine 1,3-dioxides
Zelenov VP, Voronin AA, Churakov AM, Strelenko YA, Struchkova MI, Tartakovsky VA (2013) Amino(tert-butyl-NNO-azoxy)furoxans: synthesis, isomerization, and rearrangement of N-acetyl derivatives. Russ Chem Bull Int Ed 62:117–122
Acknowledgments
The authors appreciate the 086 Project and the Research Fund for Natural Science Foundation of Jiangsu Province (NO. BK20130755) for supporting this work.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, T., Zhang, T., Xu, L. et al. Theoretical studies on vicinal-tetrazine compounds: furoxano-1,2,3,4-tetrazine-1,3,5-trioxide (FTTO-α) and furoxano-1,2,3,4-tetrazine-1,3,7-trioxide (FTTO-β). J Mol Model 20, 2516 (2014). https://doi.org/10.1007/s00894-014-2516-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2516-x