
Abstract
The cytochromes c′ (CYTcp) are found in denitrifying, methanotrophic and photosynthetic bacteria. These proteins are able to form stable adducts with CO and NO but not with O2. The binding of NO to CYTcp currently provides the best structural model for the NO activation mechanism of soluble guanylate cyclase. Ligand binding in CYTcps has been shown to be highly dependent on residues in both the proximal and distal heme pockets. Group 1 CYTcps typically have a phenylalanine residue positioned close to the distal face of heme, while for group 2, this residue is typically leucine. We have structurally, spectroscopically and kinetically characterised the CYTcp from Shewanella frigidimarina (SFCP), a protein that has a distal phenylalanine residue and a lysine in the proximal pocket in place of the more common arginine. Each monomer of the SFCP dimer folds as a 4-alpha-helical bundle in a similar manner to CYTcps previously characterised. SFCP exhibits biphasic binding kinetics for both NO and CO as a result of the high level of steric hindrance from the aromatic side chain of residue Phe 16. The binding of distal ligands is thus controlled by the conformation of the phenylalanine ring. Only a proximal 5-coordinate NO adduct, confirmed by structural data, is observed with no detectable hexacoordinate distal NO adduct.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To achieve their diverse biological functions, heme proteins face the challenge of discriminating between the similarly sized diatomic gas ligands NO, CO and O2. The cytochromes c′ (CYTcp) are mono-His ligated, periplasmic class II c-type cytochromes, widely distributed among denitrifying, methanotrophic and photosynthetic bacteria [1, 2]. They are typically homodimers with each monomer forming a four-α-helix bundle and containing a pentacoordinate (5c) c-type heme with a proximal histidine ligand and a vacant distal coordination site. Although the exact physiological functions of CYTcp remain unclear, previous studies have suggested roles in suppression of toxic levels of NO [3], defence against nitrosoative stress or an NO reductase activity [4], or NO shuttling between nitrite reductase and NO reductase during denitrification [5].
CYTcps have been classified into two groups based on the nature of their distal heme pockets [6–8]. Group 1 CYTcps, including those from Rhodobacter capsulatus (RCCP) and Chromatium vinosum (CVCP), possess an aromatic residue (Phe or Tyr) above the distal heme coordination site. In contrast, group 2 CYTcps such as that from Alcaligenes xylosoxidans (AXCP) have an aliphatic residue (Leu, or rarely Met) in this position. The best-characterised CYTcp to date is AXCP, where the ferrous protein reacts with NO to form a predominantly 5c proximal heme–nitrosyl complex via a 6c distal NO intermediate [9–11], forms only a hexacoordinate (6c) distal CO complex and does not form a stable complex with molecular oxygen [9]. In contrast, the group 1 RCCP and CYTcps from several other species form an equilibrium mixture of 6c and 5c heme–nitrosyl species in varying proportions [12]. The conserved hydrophobic distal pocket residues play a major role in the fine tuning of exogenous ligand binding and discrimination, since the mutation of distal Leu 16 in AXCP dramatically affects the distal heme affinity for diatomic gas ligands and the preference for 6- or 5-coordinate binding [13, 14]. In contrast, mutation of the proximal pocket Arg 124 in AXCP resulted in accelerated formation of the final, proximal NO species.
A particular interest in the ligand binding and discrimination properties of CYTcp stems from similarities with the activation mechanism of soluble guanylate cyclase (sGC), including the formation of a 5cNO proximal heme adduct [15, 16]. In the absence of a crystal structure of the sGC heme domain, mechanistic findings on CYTcp offer insights into the activation of sGC by NO. A proximal 5cNO species has also been reported in the pro-apoptotic cytochrome c/cardiolipin complex, again with no structural characterisation yet available [17]. Very recently, a proximal NO-binding mode has been structurally characterised in an H-NOX [18]. Despite the characterisation and structure determination of several CYTcps, structural data for NO complexes are available only for AXCP and its mutants. Here, we describe the structural, biochemical and kinetic characterisation of CYTcp from the cold water, Gram-negative bacterium Shewanella frigidimarina (SFCP). SFCP has an unusual combination of distal (Phe) and proximal (Lys) heme pocket residues. Stopped-flow and laser flash photolysis kinetic data revealed the formation of a distal 6cCO adduct in SFCP and accelerated formation of a 5c proximal NO species without any detectable 6c intermediate. The NO-bound SFCP structure presented here is only the second structure of any CYTcp–NO complex and the first structure for a nitrosyl complex of a group 1 CYTcp. Both CO- and NO-binding kinetics are biphasic, indicative of a conformational element in the mechanism. We propose a novel mechanism where the binding of gaseous ligands to the distal face of the heme is conformationally gated by a noncoordinated Phe side chain.
Materials and methods
Protein expression and purification
The gene encoding SFCP (accession number Q07Z15), including the signal peptide was amplified from S. frigidimarina genomic DNA using Taq DNA polymerase (forward primer: gtgaagggaattcatgaaaaaaatattac; reverse primer: tcagtagcaaggatccttagtcttttttg), subcloned into pGEM-T easy and cloned into the expression vector pMMB503EH [19] to generate plasmid pAG2. E. coli XL-1 Blue cells (Stratagene USA) were used for general cloning, and were routinely grown at 37 °C in Luria–Bertani (LB) medium or plates. When required, antibiotics were used at the following final concentrations: streptomycin (50 µg ml−1), chloramphenicol (20 µg ml−1). E. coli BL21(DE3) (Novagen) competent cells containing pEC86 [20], a plasmid carrying chloramphenicol resistance and the ccmA-H genes from E. coli necessary for the maturation of c-type cytochromes, were transformed with pAG2. Cells were grown in LB or terrific broth (TB) and shaken at 220 rpm. For protein production, IPTG was added in mid-log phase to a final concentration of 119 µg/ml and the temperature lowered from 37 to 25 °C. Ferriprotoporphyrin IX chloride was added to a final concentration of 10 µg ml−1 after 5 h and 1 ml of a metal ion mix (2 mM Ni2+, 2 mM Co2+, 10 mM Zn2+, 10 mM Mn2+ and 50 mM Fe3+) after 24 h in a procedure adapted from [21].
Cultures were grown for 96 h and cells harvested by centrifugation for 20 min at 4 °C and at 6000 rpm. Harvested cells were resuspended in 20 mM Tris–HCl pH 8, and disrupted by 2 passages through an EmulsiFlex (12,000 psi). The crude extract was prepared by centrifugation at 18,000 rpm for 30 min and loaded onto a DEAE-Sepharose Fast Flow anion-exchange column previously equilibrated with two column volumes of 20 mM Tris–HCl, pH 8. SFCP was eluted using 50 mM NaCl and was further purified to homogeneity by gel filtration using a Sephadex G75 column. The purity of the protein was determined by SDS-PAGE analysis and by UV–Vis spectroscopy. A final yield of 7.5 mg of SFCP per litre of culture was obtained with a purity index of 4.44 (A410/A280).
Spectroscopy and ligand-binding studies
UV–visible absorption spectra were recorded using a Varian Cary 50 spectrophotometer at 22 °C and protein concentrations estimated using ε values for the Soret band of 80 mM−1 cm−1 for oxidised CYTcps [22]. Reduction to the ferrous state was achieved by the addition of an excess of sodium dithionite or sodium ascorbate. Oxidation was achieved by addition of 50 μl of 500 mM potassium ferricyanide to an equal volume of protein solution, with the oxidant removed from the sample immediately using a PD-10 column. To prepare CO-bound ferrous SFCP, the pre-reduced sample (in a sealed 1-ml quartz cuvette) was bubbled with CO gas for 2 min. Similarly, to prepare the NO-bound ferrous SFCP, 10 μl of ProliNONOate [23] stock solution (37.5 mM) (Cayman Chemical) was introduced with a Hamilton syringe into the cuvette to provide an excess of the ligand.
Electron paramagnetic resonance (EPR) spectra were measured using a Bruker EMX spectrometer equipped with a spherical high quality Bruker resonator SP9703 and an Oxford Instruments liquid helium cryostat. The g values were obtained using the built-in microwave frequency counter. Protein samples were 100 μM in 25 mM TRIS pH 7.5. NO complexes were prepared anaerobically by the addition of 1.5 mM ProliNONOate to dithionite-reduced, nitrogen-flushed SFCP samples. Syringes, needles and EPR tubes were flushed with nitrogen before use. Immediately following the NO-donor addition, 200 μl aliquots were drawn into EPR sample tubes and flash-frozen in methanol kept on dry ice. Once frozen, the samples were transferred to liquid nitrogen and stored until use. Experimental conditions were as follows: microwave frequency 9.466 GHz, microwave power 0.0505 mW, modulation amplitude 3 G, time constant 40.96 ms, scan rate 4.77 G/s, number of scans per spectrum = 1.
Protein crystallisation and X-ray crystallography
Crystals of SFCP were grown using the hanging drop vapour diffusion method. Brown, needle-like crystals were obtained at 294 K after 2–4 days using 2 μl of 20 mg/ml protein in 20 mM Tris–HCl, pH 7, with an equivalent volume of reservoir solution containing 0.1 M HEPES pH 7 and 2.2 M ammonium sulphate. Crystals were reduced by transfer to a degassed anaerobic reservoir solution containing 100 mM ascorbate for 30 min. To prepare NO-soaked crystals of SFCP, ProliNONOate (50 µl of 50 mM stock) was injected into sealed, degassed 3-ml reservoirs containing reduced crystals (prepared as described above) for 1 h. Prior to X-ray data collection, crystals were cryoprotected by transfer to reservoir solution with 15–25 % (v/v) glycerol and flash-frozen in liquid nitrogen.
X-ray diffraction data were collected at beamline X10SA, Swiss Light Source, using a Pilatus 6M-F detector (Dectris) and an X-ray wavelength of 0.9 Å. Data were indexed in XDS [24] and scaled in Scala [25] from the CCP4i suite. The structures were solved by molecular replacement in MOLREP [26] with a monomer of the 1.45 Å-resolution crystal structure of recombinant native AXCP [Protein Data Bank (PDB) code 2YLI] [13] as the search model. Structures were refined by maximum likelihood methods using REFMAC5 [27]. Anisotropic temperature factors were refined and riding hydrogens were added to the model. Water molecules were modelled in Coot [28]. Model building between cycles of refinement was performed in Coot, and the quality of the structures was monitored using the MOLPROBITY [29] and JCSG Quality Control servers. Data collection and refinement statistics are shown in Table 1. Coordinates and structure factors were deposited in the RCSB Protein Data Bank with accession numbers 4ulv and 4cx9.
Kinetic analysis
The reduction of 5 μM ferric SFCP was achieved with a few crystals of sodium dithionite. The transient kinetics of NO binding to ferrous SFCP were monitored at 22 °C with an Applied Photophysics SX-20 stopped-flow spectrometer. The dead time of the instrument (1.3 ms) was measured by extrapolation back to time zero from CO combination with myoglobin at constant myoglobin concentration and selected CO concentrations [30]. One syringe of the stopped-flow apparatus was filled with degassed 50 mM Tris–HCl (pH 7) buffer to which 10 μl of ProliNONOate stock solution was added, while 34 μl of ferric SFCP in the same buffer was loaded into the other syringe. The final concentration of SFCP was 2.5 μM for each measurement. After the sample had been mixed, the reaction was monitored from 300 to 700 nm for 0–500 s using a photodiode array. Exponential time courses were fitted using a least-squares fitting method to yield the pseudo-first-order rate constant at each [NO] and the spectrum at each time point was deconvoluted into the component spectra of reduced and NO-bound SFCP.
Pseudo-first-order rate constants for binding were plotted against [NO] to yield the second-order rate constant. Pro-K Global Analysis Software was used to analyse NO-binding kinetics.
The CO dissociation rate constant, k off, was determined by ligand replacement with NO, while the NO dissociation rate constant was determined using dithionite as an NO scavenger. Typically, the 6cCO SFCP complex (7 μM protein and 25 µM CO solution) was mixed with a syringe containing 0.25 mM NO in buffer. Release of CO was monitored by the rate of disappearance of the 417 nm 6cCO peak or the appearance of the heme-NO absorption peak. Rate constants were insensitive to variations in the concentration of ProliNONOate (0.25–2.15 mM). To determine the NO off rate, the 5cNO SFCP complex (5 µM protein + 100 µM ProliNONOate) was mixed with 12.5 mM sodium dithionite and the release of NO was monitored by the rate of disappearance of the 398 nm 5cNO peak or the appearance of the ferrous heme absorption. The rate constants were independent of the sodium dithionite concentration (12.5–50 mM).
Flash photolysis measurements
The CO/NO association kinetics were measured at room temperature with a nanosecond laser flash photolysis system (LKS.60 from Applied Photophysics Ltd., Leatherhead, UK). In this system, the 532 nm output (∼10 ns, 110 mJ) from a Nd:YAG laser (Brilliant B from Big Sky Laser Technologies, Inc.) was employed as the photolysis beam. The output from a 150 W xenon arc lamp, at right angles to the photolysis beam, was used as the probe and was detected by a photomultiplier tube (PMT; model 1P28, Hamamatsu Corp.). The signal from the PMT was transferred to a digital oscilloscope (Infinium, Agilent Technologies) and then to a personal computer for subsequent analysis. The protein was reduced with 1 mM sodium ascorbate plus 2 µM ascorbate oxidase (Sigma), and the spectrum was recorded using the reduced protein as reference. The buffer was 50 mM Tris–HCl pH 7 and the protein concentration was 5 µM. ProliNONOate was added to the fully reduced protein to a concentration of 1 mM NO, and the spectrum recorded using the reduced protein as reference. CO was added to the ferrous protein to a concentration of 500 µM, and the spectrum was recorded using the reduced protein as reference.
Results
Purification and spectroscopic analysis
Recombinant SFCP was heterologously expressed in E. coli and purified to homogeneity. SDS-PAGE indicated a molecular weight of ~14 kDa per monomer of the functional dimer, consistent with previously characterised CYTcps. Ferric SFCP exhibited a typical UV–visible absorption spectrum with a Soret band at 404 nm, a broad β peak and a charge-transfer band at 640 nm, Fig. 1a. On reduction to the ferrous form, the Soret band shifted to 426 nm (with a shoulder at 436 nm) and the β peak occurred at 560 nm, Fig. 1a. The spectrum of ferrous SFCP exposed to excess NO was characteristic of proximal 5 coordination with a Soret band at 398 nm and very similar to the spectrum of the previous characterised 5cNO complex in AXCP [9]. The spectrum of the ferrous SFCP–CO complex featured a sharp Soret band at 417 nm and well-resolved β and α bands at 537 and 564 nm, indicative of 6-coordinate binding, i.e. CO–Fe–His (Fig. 1a). Electron paramagnetic resonance (EPR) spectra of ferrous-NO SFCP at 10 K showed a characteristic three-line hyperfine signal, again consistent with 5-coordinate NO binding to ferrous heme (Fig. 1b). The signal was very similar to that observed for 5cNO AXCP suggesting similarity between the adducts in the two proteins.

Spectroscopy of SFCP: a UV–visible absorption spectra of ferric (black), ferrous (blue), NO-bound (orange) and CO-bound (red) SFCP at pH 7. b EPR spectra of ferrous NO-bound SFCP and AXCP in the g ~ 2 region. The EPR spectrum line shapes of both proteins, exhibiting 16 G separated three lines around g = 2.013, are indicative of 5cNO binding
X-ray crystal structures
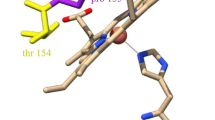
Crystal structures were determined to resolutions of 1.29 and 1.43 Å for the ‘as-isolated’ and ferrous NO-bound forms of SFCP, respectively (Table 1). Extensive attempts to obtain a structure of the 6cCO adduct were unsuccessful, likely due to the low affinity of SFCP for this ligand. The crystallographic asymmetric unit contains a homodimer, with each 128-amino acid monomer folding as an elongated left-twisted four-α-helical bundle (Fig. 2). The helices are connected by two short loops and one long loop, which are located on opposite poles of the molecule. The c-type heme is covalently bound to the Cys 119–X-X-Cys 122–His 123 motif close to the C terminus. The overall protein fold and dimeric arrangement are similar to that previously determined for other CYTcps. The SFCP dimer has the characteristic X shape found in other reported CYTcps and is stabilised by several inter-monomer interactions (supplementary figure S1). The heme environments are shown in Fig. 3a, c, for the as-isolated and NO-bound structures respectively, with corresponding structural parameters listed in Table 2.

The overall crystal structure of as-isolated SFCP with the two monomers of the dimer coloured red and green. The heme groups are represented in blue (monomer A) and orange (monomer B) respectively. Each monomer folds as a 4 α-helical bundle

Heme site structures. a 2Fo–Fc electron density map, contoured at 1σ showing the heme environment in as-isolated SFCP; b super position of the heme environment of SFCP (blue), RCCP [34] (red) and RGCP [31] (green). Residue positions that have the same amino acid are labelled in black, otherwise following the above colour scheme; Fe atoms are shown as spheres. Note that a solvent-filled channel from the distal pocket to bulk solvent is present in RCCP but in SFCP is blocked by the side chain of Met 85 and in RGCP by Leu 56; c 2Fo–Fc electron density map, contoured at 1σ showing the heme environment for the proximal NO complex of SFCP (monomer A); d superposition of the SFCP proximal 5cNO structure in monomer A (blue) with that in native (red), R124K (green) and R124F (magenta) AXCP. Note that residue 124 is omitted for clarity
The distal heme pocket is buried in the protein interior while the proximal pocket is partially exposed to solvent. The distal pocket is predominantly hydrophobic with many aromatic residues (Phe 16, Phe 59, Phe 81 and Phe 88), and contains no water molecules, while the proximal pocket contains hydrophilic residues (including the proximal heme ligand His 122 and charged residue Lys 126), which interact with several water molecules. Surprisingly, no clear solvent accessible channel between bulk solvent and the distal pocket is found in the structure, since such channels were a previously observed feature of group 2 CYTcps [31]. PDBeFold analysis confirmed that the overall fold of an SFCP monomer is very similar to those of several previously characterised CYTcps, with R. gelatinosus CYTcp [32] being the closest match with a Q-score of 0.83 while AXCP structures also provided a close match with typical Q-scores of ~0.80. The heme Fe in the as-isolated SFCP structure is 5-coordinate with a proximal His 122 ligand.
The distal pocket residue Phe 16 occupies a position some 3.3 Å from the Fe atom, with its ring lying approximately parallel to the heme, such that it is likely to sterically hinder the binding of exogenous ligands to the vacant distal heme face. The Fe–His 122(Nɛ2) bond length is 2.17/2.18 Å in monomers A and B of the SFCP dimer, respectively (Table 2). The crystal was grown from protein in the ferric state, but is likely to have become reduced to the ferrous form due to X-ray exposure, as previously demonstrated for several heme proteins [33, 34]. A superposition of the heme environments in the as-isolated SFCP structure with that from RCCP and RGCP is given in Fig. 3b. The positive charge in the proximal pocket typically provided by an Arg residue is maintained in SFCP by residue Lys 126 and at a similar distance from Fe. The crystal structure of the 5cNO complex of SFCP was determined to a resolution of 1.43 Å. In monomer A, a NO molecule is bound at the proximal face of heme (Fig. 3c) with a Fe–N bond length of 1.77 Å and a Fe–N–O angle of 128°. The displaced ligand His 122 forms a hydrogen bond with Asp 123, in a similar manner to AXCP. The environment of the proximal pocket differs from that of AXCP in that a Lys residue occupies the position of residue 124 in AXCP. Lys 126 does not form bonds to either NO or His 120. The remainder of the proximal pocket is similar to that of AXCP. Interestingly, in monomer B, a glycerol molecule (from the cryoprotectant solution) within the proximal pocket interacts with NO which is bent away from the glycerol, resulting in Fe–N and Fe–N–O values of 1.91° and 115° respectively (supplementary figure S2). Such an interaction will not occur in solution spectroscopy where glycerol is absent or indeed in vivo, but indicates the extent to which Fe–N–O geometry can be influenced by steric effects. A superposition of the SFCP 5cNO complex with that in native AXCP is shown in Fig. 3d. The Fe–N–O angle is significantly smaller in SFCP than AXCP. Ligand geometries are summarised in Table 2.
Kinetics of NO binding
On rapidly mixing ferrous SFCP with excess NO, an optical transition was observed (Fig. 4) that reported the bleaching of the ferrous 5c species (absorption maximum at 427 nm) and the simultaneous appearance of a 5cNO adduct of the heme (with absorption maximum at 395 nm). No intermediate with an absorption max at ~416 nm, characteristic of 6cNO species in other CYTcps, was observed for SFCP. The time course of this transition could be fitted to a single exponential, the rate constant of which was linearly dependent on [NO], yielding a second-order rate constant for NO binding of 6.89 (±0.26) × 103 M−1 s−1 (supplementary figure S3a). To ascertain whether the intense white light, employed using the diode array, perturbed the binding kinetics, this rate constant was also determined employing monochromatic, less intense light, and a photomultiplier system and found to be essentially unchanged. In contrast, the NO dissociation rate constant was found to be strongly influenced by the white light of the diode array system, which strongly enhanced the dissociation of the bound ligand. Thus, the NO dissociation rate constant was determined using attenuated monochromatic light and a photomultiplier system by mixing the CYTcp NO adduct with sodium dithionite, which acts as an NO scavenger. The rate constant was found to be 9.16 (±2.9) × 10−5 s−1 and independent of the dithionite concentration over the range 12.5–50 mM as opposed to a ~150-fold faster rate (1.63 × 10−2 s−1) in the diode array (data not shown).

Stopped-flow absorption measurements of the reaction of ferrous SFCP with 2.15 mM NO at 22 °C. a Time-dependent absorption measurements with a split time base between 0.0012 and 0.492 s. b The 427-nm time course overlaid with a kinetic fit (red) for an A to B reaction. Spectra recorded between 0 and 1 s after mixing show a build-up of absorption near 395 nm at the expense of the peak near 427 nm, along with an isosbestic point near 409 nm, consistent with conversion to the 5cNO end product. c The calculated absorption spectra of the kinetic components of the reaction. d The time-dependent populations of the calculated absorption spectra
Exposure of the ferrous NO complex of SFCP to laser illumination (~10 ns flash) led to the rupture of the NO–iron bond. Following photolysis, NO recombined with the heme in a biphasic time course that could be fitted to the sum of two well-resolved exponentials (supplementary figure S3b). The total amplitude and sign of the signal were wavelength dependent and consistent with the transition from 5c ferrous to 5cNO species and with the static difference spectrum, measured from UV–Vis data, for this transition. Moreover, the difference spectrum associated with each phase of the time course could similarly be adequately fitted to the static difference spectrum (Fig. 5). The rate constants describing each exponential dependence were linearly dependent on [NO] (supplementary figure S3c) yielding second-order rate constants for the two processes of 1.42 (±0.25) × 105 M−1 s−1 and 3.82 (±0.42) × 103 M−1 s−1, the latter being in reasonable agreement with that determined by stopped-flow spectroscopy (Table 3).

Correspondence of the static (line) and flash photolysis-derived (red dots) difference spectra between the 5cNO and 5c ferrous species in SFCP. a Corresponds to the fast phase and b corresponds to the slow phase
Kinetics of CO binding
Combination of CO with ferrous SFCP in stopped-flow experiments was so strongly influenced by white light that it proved impossible to monitor the process using the diode array system. Thus, the kinetics of combination was determined using attenuated monochromatic light and a photomultiplier system. On mixing the ferrous protein with CO a monophasic, exponential time course was observed at all wavelengths. The amplitude and sign of the signal was consistent with transition from a 5c ferrous form to a 6cCO adduct, as expected from the static spectrum, and the observed rate constant was linearly dependent on [CO] (Fig. 6) yielding k on = 53.6 (±2.97) M−1 s−1 and k off = 1.46 (±0.09) × 10−2 s−1. Together, these provide an estimate of the dissociation equilibrium constant K D of 272 (±31) µM. This value may be compared to that obtained from the dependence of the amplitude of the signal, at a given wavelength, on [CO] (Fig. 6b). This provides a K D value of ~203 (±13) µM in reasonable agreement to that determined from the rate constants. Subsequent to laser flash photolysis, CO recombination followed essentially a double exponential time course (see later and supplementary figure S4). The spectra associated with each of these two major phases were identical in line shape to the static difference spectrum between the 6cCO and 5c ferrous adducts, confirming that the bleaching of the CO-bound species is concurrent with the production of the 5c ferrous species. The ratio of the relative amplitudes of the two major phases (fast to slow) increases with increasing [CO]. The rate constants of both phases were [CO] dependent. The second-order rate constant of the fast phase was found to be 2.19 (±0.08) × 104 M−1 s−1. The slower phase, although adequately fitted to a single exponential did contain a smaller, faster component. The slower portion, comprising the majority of this transition yielded a second-order rate constant of 48.5 (±14.0) M−1 s−1, close to that determined by stopped-flow spectroscopy (Table 3).

Stopped-flow data for the reaction of ferrous SFCP with CO. a Dependence of the pseudo-first-order rate constant for CO binding (average determined from measurements at 435 and 417 nm) on [CO]. The straight line is a fit to this data and is consistent with the standard form for second-order reactions conducted under pseudo-first-order conductions namely \(k_{\text{obs}} = k_{\text{on}} \left[ {\text{CO}} \right] + k_{\text{off}}\). b Absorbance change determined as an average of amplitude measurements at 417 and 435 nm plotted as a function of [CO]. The line shows a hyperbolic fit to this data that uses the maximum amplitude expected at each wavelength taken from static measurements. Note that the maximum absorbance change derived from the fit at infinite [CO] is 0.167
Discussion
The crystal structure of SFCP
The overall structure of each SFCP monomer is similar to that of previously described CYTcps with solvent exposed proximal and hydrophobic distal heme pockets. The SFCP structure is, in keeping with the majority of CYTcps, a homodimer. In contrast, the CYTcp from R. palustris is unique in being observed only as a monomer while R. capsulatus CYTcp has been reported to occupy a monomer–dimer equilibrium [22].
CYTcps have previously been divided into groups 1 and 2, with those in group 1 having an aromatic (typically Phe, or rarely, Tyr) residue in the distal pocket while group 2 have an aliphatic (typically Leu, or rarely, Met) residue at this position. Another criterion was that group 1 proteins have a solvent channel from the distal pocket to bulk solvent, allowing water molecules and ligands to access the distal pocket, while in group 2 proteins this channel is blocked. In SFCP, Met 85 blocks this channel, reminiscent of the Trp residue that blocks the equivalent channel in the group 2 AXCP, and indeed the distal heme pocket is devoid of water molecules. These observations suggest that at least in this respect, SFCP is more similar to a group 2 CYTcp, whereas its distal Phe would place it within group 1. We note that CYTcps have been further classified according to the identity of their basic proximal pocket residue with group (i) having Arg and group (ii) having Lys, although the assignment of proteins to groups is not consistent with that based on the distal residue and pocket criteria of Kassner [7].
Crystal structures of several CYTcps are available with residues Leu, Phe, Met or Tyr providing steric hindrance to distal ligand binding. Of these, the group 1 proteins from R. capsulatus (RCCP; PDB 1cpq [35]), R. sphaeroides (RSCP; PDB 1gqa [36]) and R. gelatinosus (RGCP; PDB 2j8w [32]) have a Phe residue in the distal pocket in the same manner as SFCP. A superposition of the heme environments in several of these structures is given in Fig. 3b. The Phe residue lies in a rather similar position in RSCP, RGCP and SFCP, parallel to the heme plane. In RCCP, the Phe orientation is marginally different to that in the other proteins, suggesting that the degree of steric hindrance may be different to that in these other proteins. In previous work [22], RCCP has been shown to exhibit an equilibrium mixture of 6c- and 5cNO complexes. Why then, does SFCP show only a proximal 5cNO adduct? One possibility is the influence of residues in the proximal heme pocket. RCCP has an Arg residue in place of the Lys 126 present in SFCP. An equivalent Arg–Lys mutation in AXCP [21] caused a small increase in k 6-5 whereas mutation to other amino acids produced larger increases, in such cases sufficient that the distal 6cNO intermediate was not observed. It is conceivable that the different proximal pocket environments in these variants and indeed in SFCP promote faster proximal histidine dissociation and formation of the 5cNO adduct. Another possibility is the nature of any channel that exists between bulk solvent and the distal pocket. This hypothesis will be addressed in future site-directed mutagenesis studies.
Whether a distal or proximal nitrosyl adduct, or indeed an equilibrium mixture of the two is observed in a particular CYTcp appears to depend on several factors, of which a dominant effect may be a balance in the NO affinities of the two heme faces. Notably, mutations of distal pocket Leu 16 in AXCP to Ala or Gly resulted in a greatly increased affinity and a trapping of the 6cNO distal species [13, 14]. Conversely, mutations in the proximal pocket of AXCP resulted in more rapid conversion to the proximal 5cNO form, for some mutants leading to the 6c distal species no longer being observed [21, 37]. The absence of such an observable 6c distal NO intermediate in SFCP may thus arise from a lowered distal affinity as a result of the bulky Phe distal residue, an increased affinity at the proximal face or a change in the solvent accessibility of the heme pockets. We were unable to observe a CO complex in SFCP crystals. It is probable that the constraints of the crystal lattice may promote the population of the occluded form of Phe (see Scheme 1 and the discussion of kinetics below) and thus precludes the formation of a CO adduct in the crystal. We note that in AXCP, with a k on for CO of 101 M−1 s−1, k off of 0.028 s−1 and consequent K D of 2.8 × 10−4 M, a CO complex was readily observed in crystals. The SFCP crystal structure suggests that in order for CO or NO to bind at the distal face, Phe16 would have to move significantly. Such conformational rearrangements may serve to explain the observed biphasic kinetics for both ligands.

Mechanism for the reaction of ferrous SFCP with NO. Negligible reverse reactions have been omitted for clarity. PheOc refers to the conformation of Phe16 that occludes NO binding to the 6th coordination position of heme. PheAc is a different Phe16 conformation where ligand access to heme is not occluded. Note that k on (the rate of binding of NO to the distal heme face in the ferrous 5c protein to yield a 6c distal intermediate) and k 6-5 (the rate of conversion of this intermediate to the final proximal NO adduct) observed in other CYTcp proteins are not distinguished in SFCP because k 6-5 ≫ k on. In SFCP, we thus observe an apparent binding constant for formation of proximal 5cNO from ferrous 5c protein
A mechanism for NO binding to AXCP has previously been proposed [11] where an initial, intermediary, 6-coordinate distal complex is formed (k on) prior to conversion of this 6c species to a final proximal 5cNO adduct (k 6-5). For AXCP, k on was reported as 4.3 × 104 M−1 s−1 with k 6-5 of 1.14 × 104 M−1 s−1 [11], with significant changes to these values in site-directed mutants [13, 14, 21]. In SFCP, this 6c intermediate is not observed and instead an apparent k on of 6.89 × 103 M−1 s−1 was found for the formation of the 5cNO adduct in SFCP (Table 2). This value is thus 1–3 orders of magnitude lower than the NO association rate constants for AXCP and its distal pocket mutations and 4–5 orders of magnitude lower than those observed for other 5c heme proteins such as myoglobin (table S2). This indicates that NO binding to SFCP is associated with a relatively high degree of steric hindrance, most likely arising from the Phe residue that lies over the distal binding site. In all NO-binding experiments, only the 5cNO adduct was observed (i.e. no observed hexacoordinate NO complex), even at substoichiometric NO concentrations. This implies that in SFCP, k 6-5 ≫ k on and thus the apparent k on reflects both the initial binding of NO to form a 6c species and the subsequent transition to the proximal form, k 6-5.
The kinetics of NO rebinding to ferrous SFCP following photolysis of the Fe–NO bond in the 5cNO complex may be reasonably modelled by two sequential reactions. A detailed mechanism is given in Scheme 1. In the absence of external ligands, ferrous SFCP exits in two conformational ensembles differing in the accessibility of the heme to approaching ligands, with this being controlled by the position of the aromatic group of Phe 16 in the distal pocket. The two Phe conformations, which exist in rapid equilibrium with each other, are indicated as PheOc for the ligand-occluding and PheAc for the ligand-accepting positions. The occluding form, PheOc, is the dominant species at room temperature. On mixing with either NO or CO, an initial hexacoordinate ligand adduct is formed between the ligand and the PheAc form. Thereafter, re-equilibration between forms PheOc and PheAc leads to a full saturation of the distal binding site. In the case of CO binding, the reaction then terminates, while for NO, this initial distal 6cNO complex (not detected) is followed rapidly by conversion to a proximal 5cNO product via putative distal 5cNO and dinitrosyl intermediates. This mechanism yields the observed second-order rate constant k obs which may be written as \(k_{\text{obs}} = k_{\text{on}} /\left( { 1 + K_{\text{eq}} } \right)\) where \(K_{\text{eq}} = k^{\prime}_{\text{conf}} /k_{\text{conf}}\). As K eq is large, this accounts for the very low values of k obs compared to the previously characterised AXCP where a 6cNO intermediate is observed. Comparison of k obs and k on (as determined from the fast phase of the flash photolysis data) suggests that K eq is of the order of 102, i.e. at equilibrium, only ~1 % of the protein has Phe in the permitting PheAc conformation.
After cleavage of the Fe–CO bond by flash photolysis, the non-occluded pentacoordinate form produced may either rebind to CO or decay to the occluded form. This is reflected in the two major kinetic phases seen in the CO flash photolysis data, which have identical spectra but very different rate constants. The fast phase represents binding to the permitting conformation and the slow phase requiring re-equilibration for full binding and therefore having the same rate constant as derived from the stopped-flow data (supplementary scheme 1). The observation that a third process of intermediate rate constant is present indicates that relaxation back to the state from which stopped-flow experiments begin involves populating a transient kinetic intermediate which is not further discussed here, but which has been observed previously in other heme proteins, e.g. human cytoglobin [38] and rice haemoglobin [39]. Unlike in the case of CO, photolysis of the pentacoordinate NO complex of SFCP yields a 4-coordinate heme species. Our kinetic analysis shows that the difference spectra are not that of a 4-coordinate to 5-coordinate species but are in fact of pentacoordinate His-ligated heme to the 5cNO adduct. This means that the fastest process following photolysis is His rebinding to the proximal side of heme but with Phe in the permitting position (PheAc). From here, this species either rebinds NO at the distal face (fast phase with k 1 = 1.42 × 105 M−1 s−1) or relaxes to the pre-dominant, occluding, PheOc conformation which then binds NO as reflected in the slow phase (k 2 of 3.82 × 103 M−1 s−1) which again is essentially the same value as that detected by stopped flow.
UV–visible spectroscopic and EPR data for the 5cNO complexes of SFCP and AXCP are very similar (Fig. 1) but the crystal structures reveal a significantly smaller Fe–N–O angle in SFCP than in AXCP. This distortion of the NO-binding geometry can only in part be assigned to the presence of Lys rather than Arg in the proximal pocket as this angle in SFCP is also smaller than in the structure of the R124 K variant of AXCP [37]. In fact, SFCP has the smallest angle of any proximal complex characterised where a single NO-binding mode is present (supplementary table S1). The presence of an even smaller angle in monomer B of the SFCP structure where a glycerol molecule distorts NO binding is further indicative of the soft bending potential and easy distortion of the 5cNO complex by the properties and structure of the proximal heme pocket.
In conclusion, the presence of the aromatic side chain of Phe 16 in the distal pocket of SFCP introduces a conformational element to the kinetics of gas binding. In addition, formation of the final, proximal 5cNO adduct is accelerated relative to that in AXCP. This could reflect a functional role in sequestering NO, whereas proteins which retain a population of distal NO (e.g. RCCP) would be more able to release NO when required (i.e. a role in buffering NO). Our data for SFCP provide only the second structure of a proximal NO complex (all previous structures having been of AXCP and its variants) with a significantly more compressed Fe–N–O angle than in native AXCP. The significantly different proximal pocket structure extends the range of known Fe–N–O geometries and may thus inform modelling studies of the proximal NO complex in sGC.
Abbreviations
- AXCP:
-
Alcaligenes xylosoxidans cytochrome c′
- CVCP:
-
Chromatium vinosum cytochrome c′
- CYTcp:
-
Cytochrome c′
- RCCP:
-
Rhodobacter capsulatus cytochrome c′
- RGCP:
-
Rubrivivax gelatinosus cytochrome c′
- RSCP:
-
Rhodobacter sphaeroides cytochrome c′
- SFCP:
-
Shewanella frigidimarina cytochrome c′
References
Ambler RP (1991) Biochim Biophys Acta 1058:42–47
Romao MJ, Archer AM (2006) Cytochrome c′. Handbook of metalloproteins, pp 44–54
Moir JW (1999) Biochim Biophys Acta 1430:65–72
Cross R, Aish J, Paston SJ, Poole RK, Moir JW (2000) J Bacteriol 182:1442–1447
Choi PS, Grigoryants VM, Abruna HD, Scholes CP, Shapleigh JP (2005) J Bacteriol 187:4077–4085
Dobbs AJ, Anderson BF, Faber HR, Baker EN (1996) Acta Crystallogr D Biol Crystallogr 52:356–368
Kassener RJ (1991) Biochim Biophys Acta 1058:8–12
Weiss R, Gold A, Terner J (2006) Chem Rev 106:2550–2579
Andrew CR, Green EL, Lawson DM, Eady RR (2001) Biochemistry 40:4115–4122
Lawson DM, Stevenson CE, Andrew CR, Eady RR (2000) EMBO J 19:5661–5671
Andrew CR, George SJ, Lawson DM, Eady RR (2002) Biochemistry 41:2353–2360
Yoshimura T, Fujii S, Kamada H, Yamaguchi K, Suzuki S, Shidara S et al (1996) Biochim Biophys Acta 1292:39–46
Antonyuk SV, Rustage N, Petersen CA, Arnst JL, Heyes DJ, Sharma R et al (2011) Proc Natl Acad Sci 108:15780–15785
Garton EM, Pixton DA, Petersen CA, Eady RR, Hasnain SS, Andrew CR (2012) J Am Chem Soc 134:1461–1463
Stone JR, Marletta MA (1994) Biochemistry 33:5636–5640
Martin E, Berka V, Sharina I, Tsai AL (2012) Biochemistry 51:2737–2746
Silkstone G, Kapetanaki SM, Husu I, Vos MH, Wilson MT (2010) J Biol Chem 285:19785–19792
Herzik MA Jr, Jonnalagadda R, Kuriyan J, Marletta MA (2014) Proc Natl Acad Sci 111:E4156–E4164
Michel LO, Sandkvist M, Bagdasarian M (1995) Gene 152:41–45
Arslan E, Schulz H, Zufferey R, Kunzler P, Thony-Meyer L (1998) Biochem Biophys Res Commun 251:744–747
Barbieri S, Murphy LM, Sawers RG, Eady RR, Hasnain SS (2008) J Biol Inorg Chem 13:531–540
Cusanovich MA, Gibson QH (1973) J Biol Chem 248:822–834
Smith DJ, Chakravarthy D, Pulfer S, Simmons ML, Hrabie JA, Citro ML et al (1996) J Med Chem 39:1148–1156
Kabsch W (2010) Acta Crystallogr D Biol Crystallogr 66:125–132
Evans P (2006) Acta Crystallogr D Biol Crystallogr 62:72–82
Vagin A, Teplyakov A (1997) J Appl Cryst 30:1022–1025
Murshudov GN, Vagin AA, Dodson EJ (1997) Acta Crystallogr D Biol Crystallogr 53:240–255
Emsley P, Cowtan K (2004) Acta Crystallogr D Biol Crystallogr 60:2126–2132
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X et al (2007) Nucleic Acids Res 35:W375–W383
Wilson MT, Torres J (2000) In: Spectrophotometry and spectrofluorimetry: a practical approach. Oxford University Press, pp 209–240
Andrew CR, Kemper LJ, Busche TL, Tiwari AM, Kecskes MC, Stafford JM et al (2005) Biochemistry 44:8664–8672
Benini S, Rypniewski WR, Wilson KS, Ciurli SJ (2008) Inorg Biochem 102:1322–1328
Beitlich T, Kuhnel K, Schulze-Briese C, Shoeman RL, Schlichting I (2007) J Synchrotron Radiat 14:11–23
Antonyuk SV, Hough MA (2011) Biochimica et Biophysica Acta-Proteins Proteomics 1814:778–784
Tahirov TH, Misaki S, Meyer TE, Cusanovich MA, Higuchi Y, Yasuoka N (1996) Nat Struct Biol 3:459–464
Ramirez LM, Axelrod HL, Herron SR, Rupp B, Allen JP, Kantardjieff KA (2003) J Chem Crystallogr 33:413–424
Hough MA, Antonyuk SV, Barbieri S, Rustage N, McKay AL, Servid AE et al (2011) J Mol Biol 405:395–409
Beckerson P, Reeder BJ, Wilson MT (2015) FEBS Lett 509:507–512
Hargrove SM (2000) Biophys J 79:2733–2738
Acknowledgments
D.K. is supported by a School Studentship at the University of Essex. X-ray diffraction data were measured at the Swiss Light Source under long-term award 20111166 to M.H. and funded in part via BioStructX award 2370 to M.H. We acknowledge the assistance of Dr. Andrey Lebedev with restraint libraries in refinement and Gurpreet Jutla for molecular biology assistance. Professor Colin Andrew, Eastern Oregon University, is thanked for helpful discussions of the manuscript. This work was supported by Royal Society Project Grant RG120094 to M.H.
Author information
Authors and Affiliations
Corresponding author
Additional information
An Interactive 3D Complement page in Proteopedia is available at: http://proteopedia.org/wiki/index.php/Journal:JBIC:31.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manole, A., Kekilli, D., Svistunenko, D.A. et al. Conformational control of the binding of diatomic gases to cytochrome c′. J Biol Inorg Chem 20, 675–686 (2015). https://doi.org/10.1007/s00775-015-1253-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-015-1253-7