
Abstract
A re-investigation of the interaction with NO of the small tetraheme protein cytochrome c554 (C554) from Nitrosomonas europaea has shown that the 5-coordinate heme II of the two- or four-electron-reduced protein will nitrosylate reversibly. The process is first order in C554, first order in NO, and second-order overall. The rate constant for NO binding to the heme is 3000 ± 140 M−1s−1, while that for dissociation is 0.034 ± 0.009 s−1; the degree of protein reduction does not appear to significantly influence the nitrosylation rate. In contrast to a previous report (Upadhyay AK, et al. J Am Chem Soc 128:4330, 2006), this study found no evidence of C554-catalyzed NO reduction, either with \( {\text{C}}_{554}^{2 - } \) or with \( {\text{C}}_{554}^{4 - } . \) Some sub-stoichiometric oxidation of the lowest potential heme IV was detected when \( {\text{C}}_{554}^{4 - } \) was exposed to an excess of NO, but this is believed to arise from partial intramolecular electron transfer that generates {Fe(NO)}8 at heme II. The vacant heme II coordination site of C554 is crowded by three non-bonding hydrophobic amino acids. After replacing one of these (Phe156) with the smaller alanine, the nitrosylation rate for F156A2− and F156A4− was about 400× faster than for the wild type, though the rate of the reverse denitrosylation process was almost unchanged. Unlike in the wild-type C554, the 6-coordinate low-spin hemes of F156A4− oxidized over the course of several minutes after exposure to NO. Concomitant formation of N2O could explain this heme oxidation, though alternative explanations are equally plausible given the available data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The protein cytochrome c554 (C554) is a small (26 kDa) monomeric-soluble periplasmic protein containing four c-type hemes, whose primary function appears to be as an electron transport protein for the ammonia-oxidizing bacterium Nitrosomonas europaea [1,2,3,4,5,6]. However, in 2006, Upadhay and coworkers published a paper in which they reported that C554 was capable of binding nitric oxide and catalytically reducing it to nitrous oxide [7], which raised the prospect of an ancillary role as an NO reductase for the protein. This paper presents a detailed mechanistic investigation of nitric oxide’s interaction with C554 and with a variant of the protein.
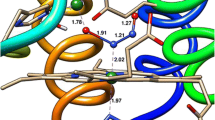
As is characteristic of c-hemes, each of the four C554 hemes is covalently attached to the protein by two thioether linkages with cysteines that are part of the amino acid sequence C–X–Y–C–H. In this motif, the histidine residue that ends the sequence is one of the heme axial ligands. Three out of four hemes in C554 are six-coordinate with bis-histidine axial ligation, while the fourth is five-coordinate with a single histidine axial ligand (Fig. 1a) [2, 3]. The three 6-coordinate hemes are all low-spin ferric in the as-isolated protein, while the 5-coordinate heme is high-spin ferric [4]. Figure 1b, c depicts the environment immediately surrounding the 5-coordinate high-spin heme. Though the iron center has a vacant site, this site is shielded by the three residues Thr 154, Pro 155, and Phe 156. These residues are not close enough to coordinate to the iron, but leave only a small hydrophobic pocket, which is not large enough to readily accommodate common heme ligands (Fig. 1c). Nonetheless, when this heme is reduced, it is the site, where NO coordination and subsequent reduction are reported to take place [7].

a C554 heme labeling scheme used throughout this article; heme II is 5-coordinate, while hemes I, III, and IV are 6-coordinate. b Vacant site of heme II, with the bulky Thr154, Pro155, and Phe156 labeled. Note that the iron is pulled out of the heme plane in the opposite direction by the histidine ligand. c Same as b, but with Thr154, Pro155, and Phe156 rendered in space-filling mode
We found C554’s purported NO reductase activity intriguing, because the heme II vacant site at which the reduction ostensibly takes place is so different from the active sites of other known NO reductases. Above all, it seemed surprising that such reactivity could be catalyzed in a heme vacant site that appears to be barely large enough to bind a single NO molecule (Fig. 1c). The classic NO reductases (NORs) catalyze the two-electron reduction of two equivalents of NO to yield N2O (Eq. 1). These enzymes have di-iron active sites with enough space for two NO
molecules to be held in close proximity, so that they can dimerize and ultimately form N2O [8,9,10,11,12]. Model studies also suggest that the di-iron motif plays a critical role in NORs [13,14,15]. Enzyme-catalyzed N2O formation at mononuclear iron centers has been documented, but the actual reaction being catalyzed in this case is quite different. Instead of the, Eq. 1, dimerization, these enzymes formally catalyze the comproportionation of hydroxylamine and nitric oxide, as shown in Eq. 2 [16]. The best studied comproportionation-mediating enzymes are cytochrome P460 from N. europaea [17, 18] and the fungal enzyme cytochrome P450nor [19, 20]. Both these
enzymes have more spacious active sites than the vacant site in C554, capable of accommodating NO and NH2OH in close proximity, as well as amino acid residues that could facilitate putative comproportionation steps [17,18,19,20].
In addition to the chemical uniqueness of C554 as a putative NO reductase, a more general incentive for studying the interaction of NO with C554 is that, for ecological reasons, it is becoming increasingly important to account for biological processes that generate N2O. The ever greater use of the Haber process to fix nitrogen for agricultural consumption has resulted in historically high accumulation of reactive nitrogen species such as \( {\text{NH}}_{4}^{ + } \) and \( {\text{NO}}_{3}^{ - } \) in the biosphere, and this is having many unintended negative consequences that will have to be mitigated in the coming years [21,22,23,24,25]. Nitrous oxide, a byproduct of denitrification, is not normally considered to be one of the “reactive nitrogen species”, because it cannot be interconverted with these; it can only be reduced back to dinitrogen. However, because of its comparative inertness, N2O can persist in the atmosphere for prolonged periods of time. This is a problem, because N2O is a potent greenhouse gas (about 300× more potent than CO2) [23], and also an ozone depleter; indeed, a recent study showed that N2O is now the single most important ozone depleting emission, and is likely to remain as such throughout the 21st century [26, 27]. Many of the current problems caused by high accumulations of reactive nitrogen species and nitrous oxide could be mitigated by more efficient use of ammonia fertilizer, and a better understanding of the interplay between the various chemical processes in the nitrogen cycle could greatly aid microbial physiologists and ecologists, and ultimately farmers, in achieving this objective.
A major challenge in studying the reactivity of NO under reducing conditions is that NO reacts directly with many commonly used reducing agents such as dithionite or methyl viologen monocation radical. This can make it difficult to deconvolute NO reduction catalyzed by a species such as C554 from the direct interaction, which is often rapid. Furthermore, except at very low concentrations, NO gas reacts rapidly with even trace oxygen, which can easily confound experimental results unless oxygen is rigorously excluded. In the past, our research group has found the light-activated NO precursor [Mn(PaPy2Q)NO]ClO4 (species 1, Scheme 1 [28], where PaPy2Q is the pentadentate ligand N,N-bis(2-pyridylmethyl)-amine-N-ethyl-2-quinoline-2-carboxamide) to be extremely useful for investigating reactions of proteins with NO [29, 30]. This species releases NO within less than 1 μs of being irradiated with a 5 ns laser pulse, and is thus ideally suited for investigating fast reactions of this gas. Furthermore, by quickly generating an aliquot of NO in situ, the method often allows the reaction of interest to be investigated before unwanted side reactions of NO, with residual oxygen or reducing agents, and can proceed to a significant extent. Herein, we re-evaluate the reactivity of NO with C554 under rigorously anaerobic conditions, exploiting the properties of 1 to minimize interference from unwanted side reactions. We also look at the interaction with NO of the F156A variant of C554.

Photolytic cleavage of 1 to produce an aquo species and free NO
Materials and methods
General materials
Common chemicals were obtained from Fisher Scientific, VWR or Sigma-Aldrich unless specified otherwise. Hexammineruthenium(II) chloride (Ru(NH3)6Cl2), hexammineruthenium(III) chloride (Ru(NH3)6Cl3), and methyl viologen were obtained from Acros. Zinc powder was from Fisher Scientific. The light-activated NO precursor 1 (Scheme 1) was prepared by the method of Eroy-Reveles et al. [28]. The pH-jump activated NO precursor 1-(N,N-diethylamino)diazene-1-ium-1,2-diolate (DEANO) was prepared as described by Drago and Paulik [31, 32]. Methods for preparing the complex K[Ru(EDTA)Cl]·2H2O and its precursors were found in articles by Wanat et al. [33], Chatterjee et al. [34], and references therein.
General instrumentation
Routine UV/Vis spectra were obtained using one of three Cary 50 (Varian) spectrophotometers available to the Pacheco group. Two of these spectrophotometers are housed in gloveboxes for obtaining spectra of air-sensitive samples. Bulk electrolysis was carried out using a BASi Epsilon EC potentiostat to set the appropriate potential. An Ag/AgCl electrode was used as a reference (BASi model RE-5B), and was periodically standardized by comparison with the methyl viologen couple [35]. Concentrations of C554 or its F156A variant, as well as the extent of the proteins’ reduction in the reaction mixtures used for stopped-flow or laser photo-initiated experiments, were assessed by UV/Vis spectroscopy using the independently obtained extinction coefficient spectra of the fully oxidized, or partially or fully reduced proteins [36].
General protein preparation
The purification protocols for C554 and its F156A variant were as described in Ref. [36]. All experiments with purified C554 and its variant were performed in solutions buffered with 50 mM N-(2-hydroxyethyl) piperazine-N-ethanesulfonic acid (HEPES) pH 7, containing 1 mM EDTA. Solutions were prepared and manipulated in a nitrogen-filled glovebox. Stock solutions were prepared daily in the glovebox, and stored in a refrigerator at 4 °C until needed.
Sample preparation for laser-initiated time-resolved spectroscopic experiments
In addition to C554 and buffer, solutions for use in laser-initiated time-resolved spectroscopy also contained a suitable reducing agent as described below, and varying concentrations of the NO-generating photosensitive species 1. Such solutions had to be prepared and kept in a darkened room at all times; a red safe-light was used to provide the minimal illumination needed to prepare the samples.
Stock solutions of two-electron-reduced C554 or F156A for laser-initiated time-resolved spectroscopy were prepared in a bulk electrolysis cell by applying potentials of − 50 mV vs SHE to solutions initially containing 200 μM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ } , \) 150 mM NaCl (as supporting electrolyte) and oxidized protein stock in standard HEPES buffer. Stock solutions containing only Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ /2 + } \) mixtures were prepared separately in a bulk electrolysis cell by applying potentials of − 50 mV vs SHE to solutions initially containing 200 μM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ } \) and 150 mM NaCl (as supporting electrolyte) in standard HEPES buffer. Finally, stock solutions of species 1 were prepared by adding aliquots of the stock 200 μM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ /2 + } \) mixtures to solid 1. Reaction mixtures for each given experiment were made by combining suitable volumes of the three stock solutions to give a total volume of 150 μL. All reaction mixtures contained the same volumes of stock C554 solution, but varying volumes of the stock 1 and 200 μM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ /2 + } \) solution.
Solutions of four-electron-reduced C554 or F156A for laser-initiated time-resolved spectroscopy were generated by gently stirring solutions initially containing the diluted oxidized protein stock, species 1 and methyl viologen (MVox, 3 μM) in a buffered suspension of zinc powder. MVox was added to the suspension to act as a mediator between the solid zinc and the dissolved protein. After 30 min, stirring was stopped, and the solid Zn was allowed to settle out in preparation for spectroscopic experiments. At this point, the methyl viologen was fully reduced to the monocation radical (MVred), as verified by UV/Vis spectroscopy.
Instrumentation for laser-initiated time-resolved spectroscopic experiments
Samples for laser-initiated time-resolved experiments were prepared in the glovebox, in 1.5 × 1.5 mm flourometer cuvettes (Starna) stoppered with greased ground glass caps. Nitric oxide was generated photochemically in the cuvettes by irradiating species 1 with 5 ns, 500 nm laser pulses from an OPO tunable laser (Opotec Rainbow Vis); NO release from the complex occurred in less than 1 μs. The laser pulse was delivered through a fiber optic cable, and was focused to a spot of 5 mm diameter that homogeneously irradiated the entire cuvette window; this arrangement minimized artifacts due to irradiated solution diffusing out of the volume being probed. The laser pulse energies were measured daily before experiments were started using a Scientech AC2501 bolometer. The laser was tuned to deliver roughly 3 mJ/pulse (with ~ 10% variability from shot to shot), which for a 5 mm spot amounted to energy intensity of about 15 mJ/cm2. Such laser pulses were found to release NO in concentrations of approximately 10% of [1] at low concentrations of 1, but the NO yield decreased progressively due to self-screening in the cuvette as [1] increased towards 1 mM and beyond [37]. The Mn product formed after release of NO is unreactive on the time scale recorded [28]. For wild-type C554, time-resolved UV/Vis spectra following the laser pulse were collected on an OLIS RSM 1000 spectrophotometer in rapid-scanning mode 1, which allowed complete spectra in the range from 362 to 587 nm to be collected at a rate of 62 scans s−1. Kinetic measurements for the very fast nitrosylation of F156A were made with the OLIS RSM 1000 in single wavelength mode, which allows spectral changes at a single wavelength to be monitored on the microsecond timescale. In all experiments, the laser pulse entered a cuvette window perpendicular to the spectrophotometer’s probe beam. Because the laser pulse irradiated the full window, the pathlength through the irradiated solution was 1.5 mm, equal to the cuvette’s pathlength.
Sample preparation for stopped-flow experiments
Stock NO for stopped-flow experiments was prepared from the precursor DEANO [31, 32, 38] in a two-step process. In the first step, a concentrated (20 mM) solution of DEANO was prepared in 0.01 M NaOH (pH 12), where it is stable for extended periods. When needed the DEANO was diluted to 200 µM with the standard pH 7.0 HEPES buffer and allowed to stand for 15 min, during which time it decomposed to yield two equivalents of NO [31, 32]. In preparation for the stopped-flow experiments, a sample of wild-type C554 or the F156A variant was two electrons reduced in the glovebox by bulk electrolysis, in solutions containing 200 µM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ } \) and 150 mM NaCl in addition to the standard HEPES buffer, as described above for the laser experiments. The two-electron-reduced protein was then nitrosylated by adding enough pH 7.0 DEANO stock to give 50 µM of NO in the reaction mixture. The final solutions contained 1.5–2 µM nitroslyated two-electron-reduced protein, and were housed in tonometers for anaerobic transport to the stopped-flow system. All other anaerobic solutions used in stopped-flow experiments were prepared by purging with nitrogen streams.
Instrumentation for denitroslyation stopped-flow experiments
Stopped-flow experiments were done on an SF-61 DX2 stopped-flow system (Hi Tech Scientific), which was made anaerobic by scrubbing the system overnight with a solution containing 2 U/ml glucose oxidase (MP biomedicals) and 1 mM glucose (Fisher Scientific). The apparatus was used in single-mixing mode, with one drive syringe containing the two-electron-reduced and nitrosylated C554 or F156A described above, and the other containing a 200 µM anaerobic solution of [Ru(EDTA)OH2]− in the standard HEPES buffer. The protein solution was rapidly mixed with the [Ru(EDTA)OH2]−, and spectral changes were monitored at 430 nm using photomultiplier tubes (Hi Tech Scientific) for 300 s for C554 and 160 s for F156A.
Data analysis
All data were analyzed using programs written within the commercially available software packages Origin version 6.0 or later (Microcal Software), or Mathcad 13 or later (PTC Software). The analysis strategies used in our laboratories have been previously described in general terms in references [38,39,40,41,42]. Strategies more specific to analyzing kinetic data, including the background for the equations appearing in “Results and Discussion”, can be found in references [29, 30, 43].
Results
The reaction of photo-generated NO with two-electron-reduced wild-type C554
Figure 2 shows the spectral changes observed after a solution initially containing 0.9 mM of the NO-generating species 1, 6 µM C554 and 200 µM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } \) was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO (Scheme 1). Under these conditions, C554 hemes I and II are reduced, while hemes III and IV remain oxidized (Fig. 1a) [4, 36, 44, 45]. The raw time-resolved UV/Vis spectra were first subjected to singular value decomposition (SVD) to determine the number of spectral components and smooth out noise [46, 47]. The SVD analysis showed that only three components were needed to faithfully reconstruct a noise-reduced absorbance matrix. The SVD-treated data were then fit to Eq. 3 using a global fitting routine (red traces, Fig. 2) [29, 30]. In Eq. 3, spectral component Λ0 is present immediately after the laser pulse (taken as t0), component Λ1 grows in exponentially in a process governed by the rate constant kobs, and component Λ2 grows in linearly with time. The data were fit to Eq. 3 with a Mathcad program that allowed kobs to be manually adjusted, after which a pseudoinverse could be calculated to generate trial values of a matrix Λ that contained the spectral components [29, 30]. The value of

a Spectral changes at selected times observed after a solution initially containing 0.9 mM of the NO-generating species 1, 6 µM C554 and 200 µM Ru(NH3) 2+6 was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO. The red dotted traces are the least-squares best fits using Eq. 3. b Changes in absorbance vs time are plotted for 415, 430 and 500 nm, where individual species make significant contributions; the red dotted traces are from the least-squares best fits using Eq. 3
kobs was adjusted until the least-squares difference between the experimental absorbance values and the matrix of ΔAλ,t values obtained from Eq. 3 was minimized.
Figure 3 shows the spectral components obtained from the fitting process. The component Λ0 (Fig. 3a) arises from the denitrosylation of species 1 within the dead time of the experiment. This component was fit with the independently known extinction coefficient spectrum of species 1, and the amount of NO generated by the laser pulse in the experiment was determined to be 31 μM from this fit. The component Λ1 (Fig. 3b) grows in exponentially, and is attributed to nitroslyation of the reduced 5-coordinate heme II of C554 [7]. Finally, component Λ2 (Fig. 3c) grows in linearly, and arises from the release of NO from species 1 caused by the spectrophotometer probe-beam irradiation during the experiment (similar linear components are seen when collecting UV/Vis time series of solutions containing only species 1) [29, 30]. The Λ2 component was fit using the independently known extinction coefficient spectrum of species 1, with an admixture of component Λ1 attributable to the extra C554 nitrosylation that accompanied the continued slow addition of NO. Small admixtures of the C554 nitrosylation spectrum Λ1 are also seen in the Λ0 component, but this is probably an artifact of the data reduction procedure, based on observations obtained by subjecting synthetic data of similar intensity and noisiness to the same procedure (including SVD analysis).

Spectral components Λ0-Λ2 generated by fitting the SVD-processed, Fig. 2, data to Eq. 3 (blue traces). The red traces were generated with the independently known extinction coefficient spectrum of species 1 and admixtures of Λ1. From the fit of Λ0 (a), one can calculate that 31 µM of NO were generated by the laser pulse in this experiment. The fit of Λ2 (c) mainly reflects the amount of 1 photolyzed per second due to the spectrophotometer probe beam; the NO so generated then nitrosylates additional C554
The analysis described in Figs. 2 and 3 was repeated with solutions containing varying concentrations of 1 and a nominally constant concentration of C554. These experiments showed that kobs varied linearly with the amount of NO generated by the laser pulses (Fig. 4). A least-squares fit of the data provided a non-zero intercept (red trace, Fig. 4), which suggests that exposure of two-electron-reduced C554 to photo-generated NO results in establishment of an equilibrium between the reduced C554 and its nitrosylated form (C5542− and C5542− NO, respectively, Scheme 2). The position and rate of establishment of this equilibrium depend visibly on the amount of NO present. Accordingly, the value of kobs at a given NO concentration will be given by Eq. 4 (Supplementary Material). Note that in all cases the amount of NO
generated by the laser pulse exceeded the amount of C554 nitrosylated by a sufficient amount to maintain pseudo first-order conditions. From the linear fit to the, Fig. 4, data, rate constant values of 3000 ± 140 M−1s−1 and 0.034 ± 0.009 s−1 were obtained for kon and koff, respectively. An equilibrium constant Keq of (9 ± 2) × 104 M−1 for the, Scheme 2, nitrosylation was subsequently calculated from the ratio kon/koff.

Plot of kobs associated with appearance of the exponential component Λ1 (Eq. 3), vs NO concentration obtained by fitting the corresponding t0 component Λ0. Red trace: least-squares fit of the data to a straight line (Eq. 4). Slope = 3000 ± 140 M−1s−1; Intercept = 0.034 ± 0.009 s−1; these are assigned as kon and koff, respectively, as defined in Scheme 2 and Eq. 4

Equilibrium between two-electron-reduced C554 and its nitrosylated form; nitrosylation of the heme II vacant site is shown schematically
The reaction of photo-generated NO with two-electron-reduced F156A
Figure 5 shows the spectral change observed immediately after a solution initially containing 900 µM of the NO-generating species 1; 5 µM F156A and 200 µM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } \) was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO (Scheme 1). As with the wild-type C554, two hemes in F156A, including the 5-coordinate heme II, are reduced under these conditions, while the other two remain oxidized [36]. In Fig. 5, spectrum shows absorbance decreases centered around 431 and 500 nm, and an absorbance increase centered around 413 nm. Least-squares fitting of this spectrum with the independently known extinction coefficient spectrum of 1, and an estimated extinction coefficient difference spectrum for wild-type \( {\text{C}}_{554}^{2 - } \) nitrosylation obtained by a procedure described in Supplementary Material, gave a plausible fit (red trace, Fig. 5). This shows that two-electron-reduced F156A nitrosylates much faster than wild type, within milliseconds instead of tens of seconds (compare with Fig. 2). Further minor spectral changes were seen in the minutes following the laser pulse (data not shown). These could be attributed to additional photolysis of 1 in the spectrophotometer probe beam and associated F156A2− nitrosylation. No evidence was seen in this experiment for F156A2− oxidation.

Blue trace: spectral change observed immediately (within 16 ms) after a solution initially containing approximately 900 µM of the NO-generating species 1, 5 µM F156A, and 200 µM Ru(NH3) 2+6 was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO. Red trace: least-squares best fit using the independently known extinction coefficient spectrum of 1 and the extinction coefficient difference spectrum associated with wild-type \( {\text{C}}_{554}^{2 - } \) nitrosylation (Supplementary Material). From the fit, one can estimate that the laser pulse photo-generated about 48 μM NO
F156A2− nitrosylation proved too fast to follow reliably with the spectrophotometer in rapid-scanning mode, so quantitative kinetic measurements were made with the instrument in fixed-wavelength mode. Figure 6 shows that the change in absorbance at 430 nm over 1 s after a solution initially containing 12 µM F156A, 200 µM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } \) and 48 µM of the NO-generating species 1 was irradiated with a 500 nm, 5 ns laser pulse. The data were well fit with a single exponential function (red trace, Fig. 6). The analysis described for Fig. 6 was repeated with solutions containing varying concentrations of two-electron-reduced F156A and a constant concentration of species 1. The values of the rate constant kobs obtained from the exponential fits varied linearly with concentration of F156A2−, as shown in Fig. 7.

Change in absorbance at 430 nm over 1 s observed after a solution initially containing 12 µM F156A, 200 µM Ru(NH3) 2+6 and 48 µM of the NO-generating species 1 was irradiated with a 500 nm, 5 ns laser pulse. Red trace: least-squares best fit to an exponential function

Blue circles: plot of kobs associated with the exponential absorbance decrease at 430 nm vs F156A2− concentration. Red trace: least-squares fit of the data to a straight line (Eq. 4). Slope = (1.23±0.09) × 106 M−1s−1; intercept = 0.5 ± 1 s−1. Note that the intercept is zero within the precision of the measurement
Presumably, by analogy with the wild-type case, F156A2− nitrosylation is reversible, as shown in Scheme 2, so that kobs should vary linearly with [NO], as shown in Eq. 4. However, the intercept in Fig. 7 is zero within the precision of the measurements, so koff cannot be obtained from this method. From the slope of Fig. 7 line, kon was estimated to be (1.23 ± 0.09)×106 M−1s−1, which is about 400× higher than the kon value obtained for the wild type (Fig. 4).
Stopped-flow investigation of C5542−NO and F156A2−NO denitroslyation
As the koff value could not be obtained from the, Fig. 7, results, an alternative strategy was developed to measure this parameter directly, using a well-known NO-trapping method [33, 34, 48]. In this method, NO released from a labile nitrosylated protein of interest is rapidly and irreversibly trapped by [Ru(EDTA)OH2]−, as shown in Scheme 3. Assuming that dissociation of NO from the protein of interest is rate limiting, the method is readily used to measure the dissociation rate constant. The method was first tested with wild-type C554, for which the dissociation rate constant had been previously determined (see Fig. 4); the denitrosylation was monitored by UV/Vis stopped-flow spectroscopy. \( {\text{C}}_{554}^{2 - } \) was first fully nitrosylated by adding an excess of NO, obtained from decomposition of the precursor DEANO [31, 32, 38], as described in “Methods”.

NO scavenging by [Ru(EDTA)OH2]−
Figure 8a shows the change in absorbance at 430 nm observed after a solution initially containing 2.1 µM wild-type \( {\text{C}}_{554}^{2 - } \)NO and about 50 μM free NO was mixed by stopped flow with a second solution containing 200 µM of the NO-trapping compound [Ru(EDTA)OH2]−. The data were fit using a two-exponential function (red trace, Fig. 8a), which gave rate constant values of 0.018 and 0.090 s−1. The amplitude associated with the slow rate constant is 4× that of the faster one and that rate constant value is comparable to the koff value obtained in the, Fig. 4, experiments (0.034 ± 0.009 s−1). The minor exponential component is believed to arise from heme oxidation, either directly or indirectly, by residual oxygen. In support of this conjecture, the UV/Vis spectrum taken after stopped-flow mixing did show that some heme oxidation had accompanied denitrosylation.

a Blue trace: change in absorbance at 430 nm observed after a solution initially containing 2.1 µM wild-type \( {\text{C}}_{554}^{2 - } \)NO was mixed by stopped-flow with a second solution containing 200 µM of the NO-trapping compound [Ru(EDTA)OH2]−. Red trace: least-squares fit of the data with a two-exponential function. b Similar to a, but syringe 1 contained 1.5 µM F156A2−NO instead of wild-type protein. In both cases, the concentration of [Ru(EDTA)OH2]− was sufficient to remove free NO as well as NO being released from nitrosylated protein
The stopped-flow experiment was repeated using F156A in place of C554 to determine koff for F156A2−NO denitrosylation (in the process analogous to the wild-type, Scheme 2, equilibrium). The blue trace in Fig. 8b shows the corresponding change in absorbance at 430 nm when 1.5 µM of F156A2−NO (pre-nitrosolyated with 50 μM NO, as described in “Methods”) was rapidly mixed with 200 µM of [Ru(EDTA)OH2]−. As before, the data were fit using a two-exponential decay equation (red trace, Fig. 8b), which in this case gave rate constant values of 0.021 and 0.070 s−1. Once again, inspection of the spectrum obtained after stopped-flow mixing showed that some heme oxidation had accompanied denitrosylation. In this case, the amplitudes associated with the two rate constants were more similar (the higher valued one had an amplitude about 1.4× larger than the lower valued one), which makes it difficult to be sure of which rate constant is associated with denitrosylation and which is tied to oxidation. However, given that the two rate constants are within a factor of 4 of each other in both the wild type and variant proteins, the salient fact is that the koff values for \( {\text{C}}_{554}^{2 - } \)NO and F156A2−NO must be in the same ballpark, whereas the kon value for NO binding to F156A2− is 400× higher than the kon value obtained for the wild type. Using the two rate constants obtained from, Fig. 8b, as lower and upper estimates of the possible koff values, one can estimate the equilibrium constant Keq = kon/koff for F156A2− nitrosylation to be about (4±2) × 107 M−1. This represents an NO affinity about 440× stronger for the F156A2− mutant compared to the wild-type \( {\text{C}}_{554}^{2 - } \), which is almost entirely due to the higher kon value for the former.
The reaction of photo-generated NO with four-electron-reduced wild-type C554
Figure 9 shows the spectral changes observed after a solution initially containing 4 µM C554, 0.75 mM of the NO-generating species 1 (Scheme 1), and 3 µM of methyl viologen reduced by zinc powder (see “Materials and Methods”) was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO (Scheme 1). Under these conditions, all four of the C554 hemes are initially reduced. SVD analysis of the data showed that four components were needed to faithfully reconstruct a noise-reduced absorbance matrix. The SVD-treated data were fit to Eq. 5 using a global fitting routine [29, 30]. In Eq. 5, spectral component Λ0 is present immediately
after the laser pulse (taken as t0), components Λ1 and Λ3 grow in exponentially in processes governed by the rate constants k1obs and k2obs, respectively, while component Λ2 grows in linearly with time. In a manner analogous to those used with the partially reduced cases, the data were fit to Eq. 5 with a Mathcad program that allowed the two first-order rate constants to be manually adjusted, after which a pseudoinverse could be calculated to generate trial values of a matrix Λ that contained the spectral components [29, 30]. The values of the rate constants were adjusted until the least-squares difference between the experimental absorbance values and the matrix of ΔAλ,t values obtained from Eq. 5 was minimized.

a Spectral changes at selected times observed when a solution initially containing 0.75 mM of the NO-generating species 1, 4 µM C554 and 3 µM MVred was exposed to a 500 nm, 5 ns laser pulse which fragments 1 to release NO. The purple traces are for the first 1000 ms, the blue traces are at 5 s intervals, and the green traces are at 20 s intervals. The red traces are the least-squares best fits using Eq. 5. b Changes in absorbance vs time are plotted for 398, 409, 420, 434, and 500 nm, where individual species make significant contributions; the red traces are from the least-squares best fits using Eq. 5
Figure 10 shows the spectral components obtained from the fitting process. As with the two-electron-reduced case, the component Λ0 (Fig. 10a) arises from the denitroslyation of species 1 within the deadtime of the experiment, and could be fit with the independently known extinction coefficient spectrum of species 1. This revealed the amount of NO generated by the laser pulse to be 66 μM for the experiment portrayed. The component Λ1 (Fig. 10b) that grows in exponentially could be fit (red trace) using the independently obtained extinction coefficient spectrum of the methyl viologen monocation radical (MVred), and showed that 3.3 μM of this species was consumed upon exposure to NO; the rate constant k1obs obtained for the process was 1.5 s−1. The component Λ2 (Fig. 10c) that grows in linearly is once again attributed to the spectrophotometer probe-beam-induced nitric oxide release from species 1, and as before, it can be fit primarily with the extinction coefficient spectrum for species 1, with some admixture of component Λ1 from the analysis of two-electron-reduced C554 (Fig. 3b, Supplementary Material).

Spectral components Λ0–Λ3 (clockwise from top left) generated from the SVD-cleaned absorbance matrix when fit to Eq. 5 (blue traces). The red traces in a and c were generated with the known extinction coefficient spectrum of species 1, and the extinction coefficient difference spectrum of \( {\text{C}}_{554}^{2 - } \)NO obtained, as described in Supplementary Material. The red trace in b was generated with the independently obtained extinction coefficient spectrum for MVred. The red trace in d was generated with the independently obtained extinction coefficient difference spectrum associated with reduction of the low-potential hemes III and IV (Fig. 1a) [36]. Subtracting the red trace from the blue one in the upper panel of d yielded the residual shown in the lower panel, which is reminiscent of the difference spectrum obtained upon nitrosylation of \( {\text{C}}_{554}^{2 - } \) (Fig. 3b)
The final component Λ3 (Fig. 10d), which grows in exponentially with a k2obs of 0.097 s−1, exhibits features characteristic of changes at the C554 c-hemes. In particular, the absorbance decreases at 554 nm and in the region 420–440 nm, and concomitant absorbance increase near 405 nm, at first sight appear to be diagnostic of straightforward c-heme oxidation. However, more careful analysis revealed that more complex changes were occurring. The decrease at 554 nm is especially characteristic of low-spin heme oxidation. However, the largest net absorbance decrease occurs at 434 nm, which is diagnostic of changes at the single high-spin 5-coordinate heme II (Fig. 1) [36, 42, 44, 49]. Qualitative consideration of the relative magnitudes of high-spin and low-spin features suggested that component Λ3 is a composite spectrum of all four hemes, and needs to be deconvoluted. To do this, the contribution at 554 nm was subtracted out completely using the previously obtained extinction coefficient difference spectrum corresponding to reduction of the two low-potential hemes III and IV (Fig. 1a) [36]. In Fig. 10d, the contribution from the low-potential heme extinction coefficient difference spectrum that resulted in elimination of the 554 nm signal is shown by the dashed red line. The residual spectrum after the subtraction is shown in the bottom panel of Fig. 10d, and is comparable to the nitroslyated spectrum of two-electron-reduced C554 (Fig. 3b). Thus, we conclude that the primary spectral change shown in Fig. 10d is due to nitrosylation of the high-spin 5-coordinate heme, just as it was when two-electron-reduced C554 was exposed to photo-generated NO. Some oxidation of the low-spin, low-potential c-hemes occurred in tandem with the nitrosylation; however, quantitative analysis shows that this amounted to only ~ 0.8 μM of the 4 μM available low-potential heme pool (which comprises two of the four C554 hemes) [36]. Assuming that the lowest potential heme IV oxidizes first, this would amount to ~ 40% oxidation of heme IV, leaving hemes I, II, and III fully reduced (with heme II of course also being nitrosylated). This is a very different result to that reported by Upadhyay et al., who saw rapid oxidation of all low-spin hemes upon exposure of fully reduced C554 to excess NO [7].
The reaction of photo-generated NO with four-electron-reduced F156A
The behavior of four-electron-reduced F156A upon exposure to NO differs from that of the wild type in several interesting ways. Figure 11 shows the spectral changes observed after a solution initially containing 7 µm F156A, 570 μM of species 1, and 3 μM of methyl viologen reduced by zinc powder (see “Methods”), was exposed to a 500 nm, 5 ns laser pulse, which fragments 1 to release NO (Scheme 1). Under these conditions, all of the F156A hemes are initially reduced. SVD analysis of the data showed that three components were needed to faithfully reconstruct a noise-reduced absorbance matrix. The SVD-treated data were fit to Eq. 3, using the global fitting routine described earlier [29, 30].

a Spectral changes at selected times observed when a solution initially containing 0.57 mM of the NO-generating species 1, 7 µM F156A and 3 µM MVred was exposed to a 500 nm, 5 ns laser pulse which fragments 1 to release NO. The dotted red traces are the least-squares best fits using Eq. 3. b Changes in absorbance vs time at the representative wavelengths stated, with the least-squares best fits using Eq. 3 overlaid as dotted red traces
Figure 12 shows the spectral components obtained from the fitting process. As with the wild-type two-electron-reduced case, a major contributor to the component Λ0 (Fig. 12a) comes from the denitroslyation of species 1 within the deadtime of the experiment; however, now the component also has a contribution from F156A4− nitrosylation, which is taking place on the ms timescale, as seen above for the two-electron-reduced protein. The component is reasonably well fit using the independently known extinction coefficient spectrum of 1, and an estimated extinction coefficient difference spectrum for wild-type \( {\text{C}}_{554}^{2 - } \) nitrosylation obtained by a procedure described in Supplementary Material (red trace, Fig. 12a).

Spectral components Λ0–Λ2 generated by fitting the SVD-cleaned, Fig. 11, data to Eq. 3 (blue traces). The red trace in a is a fit with the known extinction coefficient of species 1 and the estimated extinction coefficient difference spectrum of \( {\text{C}}_{554}^{2 - } \)NO, obtained by analyzing nitrosylation of the two-electron-reduced wild-type protein (Supplementary Material). The red trace in c was fit with the \( {\text{C}}_{554}^{2 - } \)NO extinction coefficient difference spectrum (Supplementary Material), and admixtures of the independently obtained extinction coefficient difference spectra associated with reduction of the low-potential hemes III and IV of F156A [36]
Unlike in cases encountered earlier in this work, the major contributions to the linearly increasing component Λ2 (Fig. 12c) are no longer from photolysis of 1 in the spectrometer probe beam. Instead, the major changes are now clearly due to c-heme oxidation; the absorbance decreases at 521 and 553 are particularly diagnostic of low-spin c-heme oxidation in general, and the decreases at 414 and 422 are reminiscent of the split-Soret spectral changes seen in our laboratories when low-potential hemes in C554 and its variants were electrochemically reduced [36]. With this in mind, the Λ2 component was reasonably well fit using the extinction coefficient difference spectra obtained independently for the low-potential hemes of F156A by UV/Vis spectropotentiometry [36], as well as the estimated extinction coefficient difference spectrum for wild-type C5542− nitrosylation obtained, as described in Supplementary Material, and the extinction coefficient spectrum of 1. The least-squares best fit is shown as a red overlay in Fig. 12c.
The final component Λ1 (Fig. 12b), which grows in exponentially with a kobs of 0.05 s−1, also exhibits features characteristic of low-spin c-ferroheme oxidation; in particular, the absorbance decreases at 553 nm, 522 and 422 nm, with concomitant absorbance increase at 407 nm. Recall that when four-electron-reduced wild type was exposed to photo-generated NO nitrosylation of the 5-coordinate heme, over tens of seconds was accompanied by concurrent partial oxidation of the lowest potential C554 heme. With F156A4− nitrosylation, after laser initiation occurs in ms, without attendant c-heme oxidation (component Λ0, Fig. 12a), and subsequent oxidation appears to be much slower, on the same timescale as observed for the wild type.
Discussion
The reaction of photo-generated NO with two-electron-reduced C554
The results of our experiments with two-electron-reduced C554 are inconsistent with those published earlier by Upadhyay et al. [7]. The earlier work reported immediate oxidation of heme I upon addition of excess NO, though heme II remained nitrosylated. We saw no evidence of heme I oxidation, which would have resulted in loss of signal at 420, 554, and 524 nm in the Fig. 2, difference spectra. This was true even in the presence of as much as 100 μM photo-generated NO (a 25× excess relative to the C554 concentrations). The only changes observed in Fig. 2 and analogous spectra collected with other photo-generated NO concentrations was the absorbance decrease at 432 nm and concomitant increase at 414 nm, which could be attributed to high-spin ferrous heme II nitrosylation, as reported in the earlier paper [7]. The nitrosylation process is summarized in Scheme 2; based on the EPR results of Upadhyay et al. [7], the heme II center in C5542−(NO) would be in the {Fe(NO)}7 state [50].
Our experiments were carried out in the presence of 200 μM Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } , \) so we could not immediately rule out C554-catalyzed reduction of NO to N2O by Ru(NH3) 2+6 . Only about half of RuII would potentially be oxidized by the highest concentrations of photo-generated NO (~ 100 μM), and this would poise the solution at approximately μ20 mV vs SHE (the midpoint potential for Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 3+ } . \) As the midpoint potentials of C554 hemes I and II are ~ 50 mV [36, 44, 45], very little heme oxidation would be expected, even if all of the available NO were reduced to N2O by Ru(NH3) 2+6 . To test for this possibility, we used the thermally activated NO precursor DEANO (see “Methods”) [31, 32, 38] to expose the C554/Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } , \) solutions to NO concentrations as high as 500 μM (data not shown). No C554 oxidation was observed at NO concentrations up to 300 μM. Such concentrations would have consumed all of RuII, and subsequently all C5542−, if this species had been catalyzing NO reduction by Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } . \) Hence, this possibility could be ruled out.
It should be noted that at NO concentrations higher than ~ 300 μM, we did begin to see oxidation of both hemes I and II in the two-electron-reduced C554, and this oxidation rate increased as the NO concentration was further increased (data not shown). Though such heme oxidation at very high NO concentrations could be due to direct interaction between NO and protein, the steep dependence on [NO] instead suggests the involvement of residual oxygen. In aqueous solution, NO and O2 react to ultimately give NO2−, according to the stoichiometry shown in Eq. 6, but producing strongly oxidizing intermediates on the way [51, 52]. The reaction is second order
in NO and first order in O2, which gives it a very steep dependence on [NO], much as we are seeing in the present case. In our experiments, we went to great lengths to exclude oxygen using a glovebox, submitting all stock solutions to bulk electrolysis reduction, and providing a high concentration of Ru\( \left( {{\text{NH}}_{ 3} } \right)_{ 6}^{ 2+ } \) that would tend to scrub out residual oxygen. Such precautions may explain why we saw no \( {\text{C}}_{554}^{2 - } \) oxidation at NO concentrations up to 300 μM, whereas Upadhyay et al. did [7].
\( {\text{C}}_{554}^{2 - } \) does nitrosylate at the heme II vacant site as reported earlier [7], and our work now shows that this nitrosylation is discernibly reversible (Scheme 2). Furthermore, by investigating the dependence of the nitrosylation rate kobs on NO concentration, we were able to obtain the kinetic parameters kon and koff, as defined in Scheme 2, and the associated equilibrium constant Keq = kon/koff. Keq value obtained from this analysis, (9 ± 2) × 104 M−1, is considerably lower than is typical for ferrous hemes (Table 1); for example, the value for sperm whale myoglobin is estimated to be ~ 1011 M−1 [38, 53]. Indeed, the measured Keq is more comparable to those obtained for ferric heme nitrosylations, which tend to be in the range 104–107 M−1 (Table 1) [38, 48, 54]. A low binding constant is understandable given the steric crowding of the ostensibly vacant heme II site, where the NO is binding (Fig. 1c).
Horse heart ferrocytochrome c, which will bind NO despite having no vacant heme sites available, displays a similarly low binding constant (2.9×105 M−1, Table 1) [54]. The comparison between the behavior towards NO of the two crowded hemes, that of the horse heart protein and heme II in C554, is noteworthy. The kon value for C554 is substantially higher than that for ferrocytochrome c (3000 ± 140 M−1 s−1 compared to 8.3 M−1 s−1, a factor of 360× greater), but the difference in koff values is even higher (0.034 ± 0.009 s−1 compared to 2.9×10−5 s−1, a factor of 1200× greater) [53, 54]. Unlike in the case of C554, for NO to bind to ferrocytochrome c, the iron center must first dissociate a ligand, which likely explains the comparatively low kon value. However, the much lower koff value seen for ferrocytochrome c indicates that, once bound, the Fe–NO bond is better stabilized in this protein than in C554. Indeed, the NO koff value for ferrocytochrome c is actually 10× smaller than that seen for ferromyoglobin (2.9×10−5 s−1 vs 1.2×10−4 s−1), though the far higher kon for ferromyoglobin makes the net binding much tighter for the latter [38, 53, 54]. Interestingly, NO will also bind to ferricytochrome c, with a Keq value only ~ 20× smaller than that seen for the ferrous heme (1.6×104 vs 2.9×105, Table 1) [54], whereas it will not bind at all to C554 [7]. This may reflect the fact that the Fe–NO bonds in ground-state ferric {Fe(NO)}6 species tend to be linear instead of bent, as is typical for ferrous {Fe(NO)}7 species [55,56,57], and a linear Fe–NO species may not fit in the constrained pocket of C554 (Fig. 1c). Spectroscopic and DFT studies show that the linear {Fe(NO)}6 species are best described by the resonance form [FeII(NO+)], which have strong and short Fe–NO bonds with significant double bond character. However, {Fe(NO)}6 species are typically labile, because upon deformation of the Fe–NO bond, a dissociative excited state becomes available [55, 56]. Thus, any steric factor that hinders linear Fe–NO geometry is likely to strongly suppress bonding.
The reaction of photo-generated NO with two-electron-reduced F156A
Mutating the bulky hydrophobic amino acid Phe(156) to a smaller, but still, hydrophobic alanine profoundly increased the NO binding affinity (Scheme 2) of the two-electron-reduced mutant relative to wild-type C554. Thus, the equilibrium constant Keq for NO binding went from (9 ± 2)×104 M−1 for the wild type to (4±2)×107 M−1 for F156A2−. This is understandable given that Phe(156) sits right over the vacant site of heme II, restricting access to it (Fig. 1c). Interestingly, the increased NO affinity is due almost exclusively to an increase in kon (Scheme 2), from 3000 ± 140 M−1 s−1 in the wild type to (1.23 ± 0.09)×106 M−1 s−1 in F156A. The new rate constant is now within an order of magnitude of the kon values typical of ferrous heme proteins such as myoglobin (1.7×107 M−1 s−1, Table 1) or hemoglobin (2.5×107 M−1 s−1) [54, 57, 58]. On the other hand the rate constant for NO dissociation, koff, remains virtually unchanged in F156A2−NO (~ 0.05 s−1) relative to that of the wild-type C5542−NO (0.034 ± 0.009 s−1). It appears that the less crowded pocket in F156A allows NO to more easily access and bind heme II, but that the bulky phenylalanine does not significantly destabilize the Fe–NO bond once the NO has bound metal.
We had originally speculated that NO bound to the crowded pocket in wild-type C554 might be forced to adopt an Fe–N–O bond angle more bent than is ideal in {Fe(NO)}7 species and that this would weaken the Fe–NO bond. The results presented herein suggest that, otherwise, though in the absence of structural data on the mutant, we cannot rule out the possibility that the Fe–N–O bond in F156A is still distorted, but by Thr 154 and Pro 155, which also crowd the heme II vacant site (Fig. 1c). The koff values of both wild type and F156A are both quite high compared to those of most ferroheme proteins (Table 1), and even to those of synthetic water-soluble ferrous porphyrins, which tend to fall in the range 10−4–10−5 s−1 [48, 54, 57, 58]. This might be due to the hydrophobic character of the C554 pocket, where the bound NO would have no opportunity for stabilization by H-bonding from neighboring amino acids. However, koff for NO from FeII(TPP) (TPP = tetraphenylporphyrin) in toluene has been measured at 4.17 ×T10−5 s−1, though other synthetic ferrous porphyrin complexes denitrosylate with rate constants as high as 47 s−1 in toluene [58]. These results suggest that the factors governing the C554 koff value are likely complex, and will require further investigation.
The reaction of photo-generated NO with four-electron-reduced C554
As mentioned earlier, one of the difficulties in studying reactions with NO under strongly reducing conditions is that this species reacts directly with many potential reducing agents, including the commonly used dithionite or methyl viologen monocation radical (MVred). In the experiments presented above, solid Zn powder was used as the reducing agent. On its own, this reagent was found to react very slowly with C554 (over the course of a day; data not shown); however, in the presence of a small amount of methyl viologen as mediator, C554 could be fully reduced in about half an hour. An advantage of our experimental setup, in which NO is photo-generated in less than 1 μs, is that it allowed us to deconvolute the reaction of residual MVred with NO from that of C5544− with NO. Thus, from the Figs. 9 and 10b, analyses, one can determine quantitatively that the concentration of the MVred mediator was only about 3 μM and that this was consumed within a few seconds by the photo-generated NO (with an observed first-order rate constant of 1.5 s−1). In contrast, C5544− nitrosylation and oxidation took tens of seconds (Figs. 9 and 10d), and was readily resolved from the MVred reaction.
The results presented above are once again at odds with the results published earlier by Upadhyay et al. [7]. In the earlier work, it was reported that the three low-spin hemes of \( {\text{C}}_{554}^{4 - } \) are fully and rapidly oxidized upon exposure to excess NO, with a single turnover rate exceeding 16 s−1 [7]. In our case, we observed that exposure of 4 μM \( {\text{C}}_{554}^{4 - } \) to 66 μM of photo-generated NO resulted in oxidation of only about 40% of the available low-potential heme IV, leaving the remaining hemes reduced. Furthermore, heme IV oxidation was concomitant with heme II nitrosylation, and proceeded with an observed rate constant kobs of 0.097 s−1. This rate constant is comparable to those we observed for heme II nitrosylation in two-electron-reduced C554, and considerably below 16 s−1.
Scheme 4 proposes a minimal mechanism for the partial oxidation of heme IV that accompanies heme II nitrosylation (Figs. 9 and 10d). According to this mechanism, the {Fe(NO)}7 species generated by nitrosylation of heme II is reversibly reduced to {Fe(NO)}8, or Fe(HNO)}8, by intramolecular electron transfer (IET) from the low-potential heme IV. In this scenario, the nitrosylation would be rate limiting, and occur at roughly the same rate as it does for C5542− (Scheme 2), after which the IET equilibrium would be established rapidly.

Minimal mechanism proposed for the reaction of NO with fully reduced C554. FeII and FeIV refer to the iron centers of hemes II and IV (Fig. 1a), respectively. IET, intramolecular electron transfer. It is also possible that IET is coupled to protonation of the NO moiety, giving [{FeII(HNO)}8,Fe 3+IV ]C554
The residual spectrum of Fig. 10d after the heme oxidation contribution is subtracted looks very similar to the {Fe(NO)}7 spectrum observed upon nitrosylation of \( {\text{C}}_{554}^{2 - } \) heme II (Fig. 3b). Accordingly, the, Scheme 4, mechanism requires that {Fe(NO)}7 and {Fe(NO)}8 (or {Fe(HNO)8}) have very similar spectra, which is plausible given what is known about {Fe(NO)}7 and {Fe(NO)}8 species. To our knowledge, pure {Fe(NO)}8 heme species have not been previously reported for proteins (though the protonated {Fe(HNO)}8 species has been characterized for myoglobin [59, 60]), but such species have been extensively characterized in model complexes [55]. Spectroscopic and DFT studies have shown that reduction of {Fe(NO)}7 to {Fe(NO)}8 is accompanied by minimal electronic reorganization [55, 61]. {Fe(NO)}7 has a singly occupied molecular orbital that makes a major contribution to the Fe–N σ bond, and this MO becomes doubly occupied when {Fe(NO)}7 is reduced to {Fe(NO)}8, strengthening the Fe–N bond without significantly affecting the porphyrin MOs [55, 61].
If the Scheme 4, mechanism is for the moment assumed to be correct, it can be used to extract an estimate of the midpoint potential for {Fe(NO)}7 reduction from Fig. 10d (Supplementary Material). As shown in the, Fig. 10d, analysis presented earlier, heme II nitrosylation of \( {\text{C}}_{554}^{4 - } \) is accompanied by ~ 40% oxidation of heme IV. According to Scheme 4, this would correspond to an equilibrium constant of ~ 0.67 governing IET from ferrous heme IV to {Fe(NO)}7 heme II, which in turn would translate to a cell potential of –0.010 V for net reduction of {Fe(NO)}7 heme II by ferrous heme IV. Given that the midpoint potential for heme IV is about –0.27 V [36, 44, 45, 62], we find that the midpoint potential for {Fe(NO)}7 heme II reduction would be ~ − 0.28 V according to the, Scheme 4, mechanism (Supplementary Material). This is higher than the midpoint potentials recorded for {Fe(NO)}7 reduction in model porphyrin systems in organic media, which range from − 0.4 to − 1.1 V (vs SHE) [61, 63, 64].
The reaction of photo-generated NO with four-electron-reduced F156A
As was seen for wild-type \( {\text{C}}_{554}^{4 - } , \) exposure of F156A4− to photo-generated NO results in heme oxidation as well as heme II nitrosylation. However, there are important differences between the two cases. First, the nitrosylation rate observed for F156A4− was much faster than that observed for the wild type, yet the rate of heme oxidation was comparable. Second, the extent of heme oxidation that followed exposure to NO was substantially greater for the mutant. In the case of \( {\text{C}}_{554}^{4 - } \) wild type, it was estimated that about 40% of the low-potential heme IV was oxidized at the same time as heme II was being nitrosylated. Efforts to quantify the extent of c-heme oxidation after F156A4− nitrosylation have thus far been thwarted, because we have not been able to fit one of the spectra associated with the oxidation events (spectrum Λ1, Fig. 12b). Nevertheless, it is clear from a visual comparison of Figs. 10 and 12 that the extent of c-heme oxidation is substantially greater for the mutant.
Given the greater extent of heme oxidation found for the F156A variant, we cannot at this point rule out the possibility that this variant can reduce NO to N2O, at least stoichiometrically.
Alternatively, the extra oxidation could be explained by the minimal mechanism, as shown in Scheme 5. According to this mechanism, the initial rapid heme II nitrosylation is followed by a series of intramolecular electron transfers (IETs) from ferrohemes IV, III, and I to the FeII–NO moiety. Each IET event would be fast but unfavorable on its own, but would be coupled to a slow protonation of the bound NO that would drive the reaction forward. After three successive proton-coupled IETs, NH2OH would be released, leaving the protein in a one-electron-reduced state (F156A−). This species would then bind NO to once again leave heme II in an {Fe(NO)}7 state. Based on this mechanism, the difference in reactivity between the wild type and F156A mutant could be explained as follows. First, substitution of Phe by Ala would open up the heme II region to allow more facile access for protons. Second, the heme III midpoint potential is substantially lower than that of the wild type [36], making this heme a stronger electron donor. If the low-potential hemes III and IV provide two electrons, the third would have to be provided by the high potential heme I. However, even if the last reduction step were somewhat unfavorable, it could be driven forward by the favorable re-nitrosylation of heme II in Scheme 5. Studies are now under way to conclusively identify the mechanism that leads to heme oxidation of four-electron-reduced F156A when it is exposed to NO.

One possible process by which F156A4− could be oxidized in the presence of NO; another possibility is that NO reduction results in N2O generation
Conclusions
Our results indicate that wild-type \( {\text{C}}_{554}^{2 - } \) and \( {\text{C}}_{554}^{4 - } \) will both nitrosylate at heme II; however, we saw no evidence of the previously reported C554-catalyzed NO reduction [7], either with \( {\text{C}}_{554}^{2 - } \) or with \( {\text{C}}_{554}^{4 - } \). Some sub-stoichiometric oxidation of the lowest potential heme IV is detected when \( {\text{C}}_{554}^{4 - } \) is exposed to an excess of photo-generated NO, but the simplest explanation for this observation is that of Scheme 4, whereby heme IV partially reduces {Fe(NO)}7 to yield {Fe(NO)}8.
Heme II of the two- or four-electron-reduced F156A C554 mutant has an affinity for NO that is about 400× higher than heme II in the wild type, a difference that is almost exclusively due to a greater kon association rate constant. As was the case for C5542−, the two-electron-reduced F156A mutant will not oxidize in the presence of photo-generated NO. However, unlike in the wild-type case, rapid nitrosylation of F156A4− heme II is followed by substantial oxidation of the remaining C554 low-spin hemes. Though Scheme 5 provides a possible minimal mechanistic scheme to rationalize the results, more studies will be needed to understand the observed chemistry. EPR studies of reactive intermediates, and mass spectrometric analysis of the reaction products, should be particularly useful in this regard.
Despite their lack of NO reductase activity, nitrosylated C554 and its variants display certain properties that make them worthy of further study. For example, spectroscopic studies are now under way to more fully characterize the product obtained by nitrosylating \( {\text{C}}_{554}^{4 - } \). To our knowledge, non-protonated {Fe(NO)}8 species have not previously been characterized in proteins, so if confirmed, this would be the first example. Furthermore, nitrosylated C554 and its F156A variant are unusually resistant to trans-labilization of the heme II histidine axial ligand. We see evidence of His-dissociation in F156A4− and to a lesser extent in \( {\text{C}}_{554}^{4 - } \) after exposure to NO, but such dissociation takes place over a 12 h period, which is long compared to other known protein and model systems [55, 65]. Lehnert and coworkers made imidazoles in model systems resistant to NO-induced trans-labilization by restricting their range of motion [55, 65]; the relative rigidity of the C554 scaffold likely explains this protein’s resistance to trans-labilization as well. Studies are now under way to more fully elucidate the factors that affect the rates of trans-labilization in nitrosylated C554 and its variants.
Abbreviations
- C554 :
-
Cytochrome c554
- NOR:
-
Nitric oxide reductase
- PaPy2Q:
-
N,N-bis(2-pyridylmethyl)-amine-N-ethyl-2-quinoline-2-carboxamide)
- DEANO:
-
1-(N,N-diethylamino)diazene-1-ium-1,2-diolate
- HEPES:
-
N-(2-hydroxyethyl) piperazine-N-ethanesulfonic acid
- SVD:
-
Singular value decomposition
References
Yamanaka T, Shinra M (1974) Cytochrome c-552 and cytochrome c-554 derived from Nitrosomonas europaea. J Biochem 75:1265–1273
Iverson TM, Arciero DM, Hsu BT, Logan MSP, Hooper AB, Rees DC (1998) Heme packing motifs revealed by the crystal structure of the tetra-heme cytochrome c554 from Nitrosomonas europaea. Nat Struct Biol 5(11):1005–1012
Iverson TM, Arciero DM, Hooper AB, Rees DC (2001) High-resolution structures of the oxidized and reduced states of cytochrome c554 from Nitrosomonas europaea. J Biol Inorg Chem 6(4):390–397
Upadhyay AK, Petasis DT, Arciero DM, Hooper AB, Hendrich MP (2003) Spectroscopic characterization and assignment of reduction potentials in the tetraheme cytochrome c(554) from Nitrosomonas europaea. J Am Chem Soc 125(7):1738–1747. https://doi.org/10.1021/Ja020922x
Whittaker M, Bergmann D, Arciero D, Hooper AB (2000) Electron transfer during the oxidation of ammonia by the chemolithotrophic bacterium Nitrosomonas europaea. Biochim Biophys Acta Bioenerg 1459(2–3):346–355
Kim HJ, Zatsman A, Upadhyay A, Whittaker M, Bergmann DJ, Hendrich MP, Hooper AB (2008) Membrane tetraheme cytochrome c m552 of the ammonia-oxidizing Nitrosomonas europaea: a ubiquinone reductase. Biochemistry 47:6539–6551
Upadhyay AK, Hooper AB, Hendrich MP (2006) NO reductase activity of the tetraheme cytochrome c(554) of Nitrosomonas europaea. J Am Chem Soc 128(13):4330–4337. https://doi.org/10.1021/Ja055183+
Hendriks J, Warne A, Gohlke U, Haltia T, Ludovici C, Lubben M, Saraste M (1998) The active site of bacterial nitric oxide reductase is a dinuclear iron center. Biochemistry 37:13102–13109
Wasser IM, de Vries S, Moenne-Loccoz P, Schroder I, Karlin KD (2002) Nitric oxide in biological denitrification: Fe/Cu metalloenzyme and metal complex NOx redox chemistry. Chem Rev 102:1201–1234
Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S, Shiro Y (2010) Structural basis of biological N2O generation by bacterial nitric oxide reductase. Science 230:1666–1670
Sato N, Ishii S, Sugimoto H, Hino T, Fukumori Y, Sako Y, Shiro Y, Tosha T (2014) Structures of reduced and ligand-bound nitric oxide reductase provide insights into functional differences in respiratory enzymes. Proteins 82:1258–1271
Shiro Y (2012) Structure and function of bacterial nitric oxide reductases. Biochim Biophys Acta 1817:1907–1913
Matsumura H, Chackraborty S, Reed J, Lu Y, Moenne-Loccoz P (2016) Effect of outer-sphere side chain substitutions on the fate of the trans-iron nitrosyl dimer in heme/nonheme engineered myoglobins (FeBMbs): insights into the mechanism of denitrifying NO reductases. Biochemistry 55:2091–2099
Lin Y-W, Yeung N, Gao Y-G, Miner KD, Tian S, Robinson H, Lu Y (2010) Roles of glutamates and metal ions in a rationally designed nitric oxide reductase based on myoglobin. PNAS 107:8581–8586
Abucayon EG, Khade RL, Powell DR, Zhang Y, Richter-Addo GB (2018) Lewis acid activation of the ferrous heme–NO fragment toward the N–N coupling reaction with NO to generate N2O. J Am Chem Soc 140:4204–4207
Zhu X, Burger M, Doane TA, Horwath WR (2013) Ammonia oxidation pathways and nitrifier denitrification are significant sources of N2O and NO under low oxygen availability. PNAS 110:6328–6333
Caranto JD, Vilbert AC, Lancaster KM (2016) Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. PNAS 113:14704–14709
Vilbert AC, Caranto JD, Lancaster KM (2017) Influences of the heme–lysine crosslink in cytochrome P460 over redox catalysis and nitric oxide sensitivity. Chem Sci:DOI. https://doi.org/10.1039/c1037sc03450d
Shoun H, Fushinobu S, Jiang L, Kim S-W, Wagaki T (2012) Fungal denitrification and nitric oxide reductase cytochrome P450nor. Phil Trans R Soc B 367:1186–1194
McQuarters AB, Wirgau NE, Lehnert N (2014) Model complexes of key intermediates in fungal cytochrome P450 nitric oxide reductase (P450nor). Curr Op Chem Biol 19:82–89
Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA (2008) Transformation of the Nitrogen Cycle: recent trends, questions and potential solutions. Science 320:889–892
Duce RA, LaRoche J, Altieri K, Arrigo KR, Baker AR, Capone DG, Cornell S, Dentener F, Galloway J, Ganeshram RS, Geider RJ, Jickells T, Kuypers MM, Langlois R, Liss PS, Liu SM, Middelburg JJ, Moore CM, Nickovic S, Oschlies A, Pedersen T, Prospero J, Schlitzer R, Seitzinger S, Sorensen LL, Uematsu M, Ulloa O, Voss M, Ward B, Zamora L (2008) Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 320:893–897
Canfield DE, Glazer AN, Falkowski PG (2010) The evolution and future of earth’s nitrogen cycle. Science 330:192–196
Galloway JN, Leach AM, Bleeker A, Erisman JW (2013) A chronology of human understanding of the nitrogen cycle. Phil Trans R Soc B 368:20130120
Lehnert N, Coruzzi G, Hegg E, Seefeldt L, Stein L (2015) Feeding the world in the 21st century: grand challenges in the nitrogen cycle. National Science Foundation Workshop. https://www.nsf.gov/mps/che/workshops/nsf_nitrogen_report_int.pdf. Accessed June 2017
Ravishankara AR, Daniel JS, Portmann RW (2009) Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326:123–125
Wuebbles DJ (2009) Nitrous oxide: no laughing matter. Science 326:56–57
Eroy-Reveles A, Leung Y, Beavers CM, Olmstead MM, Mascharak PK (2008) Near-infrared light activated release of nitric oxide from designed photoactive manganese nitrosyls: strategy, design, and potential as NO donors. J Am Chem Soc 130:4447–4458
Koebke KJ, Pauly DJ, Lerner L, Liu X, Pacheco AA (2013) Does the oxidation of nitric oxide by oxyMyoglobin share an intermediate with the metMyoglobin-catalyzed isomerization of peroxynitrite? Inorg Chem 52:7623–7632
Koebke KJ, Waletzko MT, Pacheco AA (2016) Direct monitoring of the reaction between photochemically generated nitric oxide and Mycobacterium tuberculosis truncated hemoglobin N wild type and variant forms: an assessment of theoretical mechanistic predictions. Biochemistry 55:686–696
Drago RS, Paulik FE (1960) The reaction of nitrogen (II) oxide with diethylamine. J Am Chem Soc 82:96–98
Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK (1991) Complexes of NO with nucleophiles as agents for the controlled biological release of nitric-oxide—vasorelaxant Effects. J Med Chem 34(11):3242–3247
Wanat A, Schneppensieper T, Karocki A, Stochel G, van Eldik R (2002) Thermodynamics and kinetics of RuIII(edta) as an efficient scavenger for nitric oxide in aqueous solution. J Chem Soc Dalton Trans 6:941–950
Chatterjee D, Mitra A, Sengupta A, Saha P, Chatterjee M (2006) [RuIII(Medtra)(H2O)] (medtra = N-methylethylenediaminetriacetate) complex—a highly efficient NO inhibitor with low toxicity. Inorg Chim Acta 359:2285–2290
Watanabe T, Honda K (1982) Measurement of the extinction coefficient of the methyl viologen cation radical and the efficiency of its formation by semiconductor photocatalysis. J Phys Chem 86:2617–2619
McGarry JM (2017) Interaction with nitric oxide of the Nitrosomonas europaea tetraheme protein cytochrome c 554, and two of its variants, in increasingly reducing environments. Ph.D., University of Wisconsin-Milwaukee, Dept. of Chemistry and Biochemistry, Milwaukee, WI
Koebke KJ (2015) Mechanistic study of heme protein-mediated nitric oxide dioxygenation using photolytically produced nitric oxide. Ph.D., University of Wisconsin-Milwaukee, Dept. of Chemistry and Biochemistry, Milwaukee, WI
Purwar N, McGarry JM, Kostera J, Pacheco AA, Schmidt M (2011) Interaction of nitric oxide with catalase: structural and kinetic analysis. Biochemistry 50:4491–4503
Cabail MZ, Moua V, Bae E, Meyer A, Pacheco AA (2007) Quantifying the photoinduced release of nitric oxide from N,N’-bis(carboxymethyl)-N,N’-dinitroso-1,4-phenylenediamine. Effect of reducing agents on the mechanism of the photoinduced reactions. J Phys Chem A 111(7):1207–1213. https://doi.org/10.1021/Jp065487y
Kostera J, Youngblut MD, Slosarczyk JM, Pacheco AA (2008) Kinetic and product distribution analysis of NO reductase activity in Nitrosomonas europaea hydroxylamine oxidoreductase. J Biol Inorg Chem 13(7):1073–1083. https://doi.org/10.1007/s00775-008-0393-4
Kostera J, McGarry JM, Pacheco AA (2010) Enzymatic interconversion of ammonia and nitrite: the right tool for the job. Biochemistry 49:8546–8553
Youngblut M, Judd ET, Srajer V, Sayyed B, Goelzer T, Elliott SJ, Schmidt M, Pacheco AA (2012) Laue crystal structure of Shewanella oneidensis cytochrome c nitrite reductase from a high-yield expression system. J Biol Inorg Chem 17:647–662
Youngblut M, Pauly DJ, Stein N, Walters D, Conrad JA, Moran GR, Bennett B, Pacheco AA (2014) Shewanella oneidensis cytochrome c nitrite reductase (ccNiR) does not disproportionate hydroxylamine to ammonia and nitrite, despite a strongly favorable driving force. Biochemistry 53:2136–2144
Arciero DM, Collins MJ, Haladjian J, Bianco P, Hooper AB (1991) Resolution of the 4 hemes of cytochrome-C554 from Nitrosomonas europaea by redox potentiometry and optical spectroscopy. Biochemistry 30(48):11459–11465
Pulcu GS, Elmore BL, Arciero DM, Hooper AB, Elliott SJ (2007) Direct electrochemistry of tetraheme cytochrome c 554 from Nitrosomonas europaea: redox cooperativity and gating. J Am Chem Soc 129:1838–1839
Press WH, Teukolsky SA, Vetterling WT, Flannery BP (2007) Numerical recipes the art of scientific computing, 3rd edn. Cambridge University Press, New York, pp 65–75
Henry ER, Hofrichter J (1992) Singular value decomposition: application to analysis of experimental data. In: Brand L, Johnson ML (eds) Meth enzymol, vol 210. Academic Press, San Diego, pp 129–192
Laverman LE, Wanat A, Oszajca J, Stochel G, Ford PC, van Eldik R (2001) Mechanistic studies on the reversible binding of nitric oxide to metmyoglobin. J Am Chem Soc 123(2):285–293. https://doi.org/10.1021/Ja001696z
Marritt SJ, Kemp GL, Xiaoe L, Durrant JR, Cheesman MR, Butt JN (2008) Spectroelectrochemical characterization of a pentaheme cytochrome in solution and as electrocatalytically active films on nanocrystalline metal-oxide electrodes. J Am Chem Soc 130:8588–8589
Enemark JH, Feltham RD (1974) Principles of structure, bonding and reactivity for metal nitrosyl complexes. Coord Chem Rev 13:339–406
Goldstein S, Czapski G (1995) Kinetics of nitric oxide autoxidation in aqueous solution in the absence and presence of various reductants. The nature of the oxidizing intermediates. J Am Chem Soc 117:12078–12084
Goldstein S, Czapski G (1996) Mechanism of the nitrosation of thiols and amines by oxygenated NO solutions: the nature of the nitrosating intermediates. J Am Chem Soc 118:3419–3425
Moore EG, Gibson QH (1976) Cooperativity in the dissociation of nitric oxide from hemoglobin. J Biol Chem 251:2788–2794
Hoshino M, Ozawa K, Seki H, Ford PC (1993) Photochemistry of nitric-oxide adducts of water-soluble iron(III) porphyrin and ferrihemoproteins studied by nanosecond laser photolysis. J Am Chem Soc 115(21):9568–9575
Hunt AP, Lehnert N (2015) Heme-nitrosyls: electronic structure implications for function in biology. Acc Chem Res 48:2117–2125
Praneeth VKK, Paulat F, Berto TC, George SD, Nather C, Sulok CD, Lehnert N (2008) Electronic structure of six-coordinate iron(III)-porphyrin NO adducts: the elusive iron(III)-NO(radical) state and its influence on the properties of these complexes. J Am Chem Soc 130:15288–15303
Ford PC, Lorkovic IM (2002) Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes. Chem Rev 102(4):993–1017. https://doi.org/10.1021/Cr0000271
Wolak M, van Eldik R (2002) To be or not to be NO in coordination chemistry? A mechanistic approach. Coord Chem Rev 230(1–2):263–282 pii S0010-8545(01)00472-6
Lin R, Farmer PJ (2000) The HNO adduct of myoglobin: synthesis and characterization. J Am Chem Soc 122:2393–2394
Murugaeson RK, Fukuto JM, Miranda KM, Farmer PJ (2010) Reactions of HNO with heme proteins: new routes to HNO-heme complexes and insight into physiological effecs. Inorg Chem 49:6283–6292
Goodrich LE, Roy S, Alp EE, Zhao J, Hu MY, Lehnert N (2013) Electronic structure and biologically relevant reactivity of low-spin {Fe(NO)}8 porphyrin model complexes: new insight from a bis-picket fence porphyrin. Inorg Chem 52:7766–7780
Kurnikov IV, Ratner MA, Pacheco AA (2005) Redox equilibria in hydroxylamine oxidoreductase. Electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry 44(6):1856–1863. https://doi.org/10.1021/Bi048060v
Choi I-K, Liu Y, Feng DW, Paeng K-J, Ryan MD (1991) Electrochemical and spectroscopic studies of iron porphyrin nitrosyls and their reduction products. Inorg Chem 30:1832–1839
Lancon D, Kadish KM (1983) Electrochemical and spectral characterization of iron mono- and dinitrosyl porphyrins. J Am Chem Soc 105:5610–5617
Berto TC, Praneeth VKK, Goodrich LE, Lehnert N (2009) Iron-porphyrin NO complexes with covalently attached N-donor ligands: formation of a stable six-coordinate species in solution. J Am Chem Soc 131:17116–17126
Acknowledgements
Major funding for this work was provided by National Science Foundation Grants MCB-1121770, MCB-1330809, and MCB-1616824. The authors are indebted to Graham Moran and Nicholas Silvaggi of the University of Wisconsin-Milwaukee for use of their stopped-flow and FPLC chromatography systems, respectively, and to John Coates from the University of California-Berkeley for providing the WM3064 cells. JMM also wished to thank her colleagues in the laboratory, particularly Karl Koebke and Matthew Youngblut, for helpful discussions and much practical advice.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McGarry, J.M., Pacheco, A.A. Upon further analysis, neither cytochrome c554 from Nitrosomonas europaea nor its F156A variant display NO reductase activity, though both proteins bind nitric oxide reversibly. J Biol Inorg Chem 23, 861–878 (2018). https://doi.org/10.1007/s00775-018-1582-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-018-1582-4