
Abstract
The enzymes nitrous oxide reductase (N2OR) and cytochrome c oxidase (COX) are constituents of important biological processes. N2OR is the terminal reductase in a respiratory chain converting N2O to N2 in denitrifying bacteria; COX is the terminal oxidase of the aerobic respiratory chain of certain bacteria and eukaryotic organisms transforming O2 to H2O accompanied by proton pumping. Different spectroscopies including magnetic resonance techniques, were applied to show that N2OR has a mixed-valent Cys-bridged [Cu1.5+(CyS)2Cu1.5+] copper site, and that such a binuclear center, called CuA, does also exist in COX. A sequence motif shared between the CuA center of N2OR and the subunit II of COX raises the issue of a putative evolutionary relationship of the two enzymes. The suggestion of a binuclear CuA in COX, with one unpaired electron delocalized between two equivalent Cu nuclei, was difficult to accept originally, even though regarded as a clever solution to many experimental observations. This minireview in honor of Helmut Sigel traces several of the critical steps forward in understanding the nature of CuA in N2OR and COX, and discusses its unique electronic features to some extent including the contributions made by the development of methodology and the discovery of a novel multi-copper enzyme.
Graphical Abstract
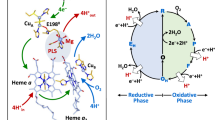
Left: X-band (9.130 GHz) and C-band (4.530 GHz, 1st harmonic display of experimental spectrum) EPR spectra of bovine heart cytochrome c oxidase, recorded at 20K. Right: Ribbon presentation of the CuA domain in cytochrome c oxidase and nitrous oxide reductase.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In this minireview, the road to our present picture of the binuclear mixed-valent copper A site, [Cu1.5+(CyS)2Cu1.5+], will be discussed from a personal point of view. The topic on the binuclear electron transfer center present in redox enzymes cytochrome c oxidase (COX) and nitrous oxide reductase (N2OR) seems well suited for honoring Helmut Sigel (University of Basel) on the occasion of his 80th birthday, in view of his long standing and deep interest in Biological Inorganic Chemistry [1,2,3,4]. It was Helmut Sigel who started the pioneering and highly regarded book series Metal Ions in Biological Sciences (MIBS) as early as 1973 [5], which is running under the title Metal Ions in Living Systems (MILS) since 2006 until today, with co-editors Astrid and Roland Sigel.
Perhaps the defining feature of the CuA center, first described in COX [6, 7], is its unusual electron paramagnetic resonance (EPR) spectrum, and for a long time a consensual explanation of the EPR properties did not exist. Notably, early on most ideas involved a combination of copper and sulfur in the CuA site. In 1974, Peisach and Blumberg described in their article entitled “Structural Implications Derived from the Analysis of Electron Paramagnetic Resonance Spectra of Natural and Artificial Copper Proteins” an interesting case of an EPR signal ascribed to copper in COX; g ║ and g ⊥ were resolved, but the hyperfine structure was not, thus the splitting must be less than 30 Gauss. This spectrum (measured at X-band) showed g values around 2.17 and 2.03, both parameters being closer to g = 2 than for any other natural or artificial copper protein. The lack of hyperfine structure was suggested to arise from a specific stereochemical distortion of electron-accepting ligands of the copper, or from a significant transfer of the unpaired spin of the copper to sulfur. Note that the Peisach–Blumberg plot applies to mononuclear Cu sites, thus the value of the hyperfine splitting (hfs) by 63,65Cu (I = 3/2) in the g ║-region (A║), by a dinuclear Cu site has to be doubled. Thus, it is not as far away from those of type 1 Cu reported in the older publications, where CuA was still considered to be a mononuclear site. The putative sulfur-free radical in this instance must be bonded to a tetrahedral carbon atom as was found in the sulfur radical (g = 2.25, 2.00, 1.98) in irradiated cysteine hydrochloride [8]. The degree of transfer of the unpaired electron spin could vary almost up to 100% and still exhibit the EPR parameters ascribed to copper in COX. Earlier, Vänngård had pointed out that, in plots of the copper hfs A║ vs g ║ there was a distinct grouping of Cu complexes or proteins of non-blue type 2 and of blue type 1 copper proteins, whereas the value for CuA, was lying way out by itself [9]. To obtain more definitive information on the ligands of CuA in COX, Chan and his associates examined the X-band EPR and electron nuclear double resonance (ENDOR) spectra of isotopically enriched (1,3-15N2-His, or ßß-2H2-Cys) COX. They concluded that there was at least one His and one Cys ligand (possibly even two of each), and they proposed a model in which a single Cu ion was ligated by two His and two Cys in tetrahedral coordination, often referred to as the Chan model (Fig. 1) [10,11,12].

Structure for the CuA center in COX proposed by Chan and associates [10]
In 1980, Mims et al. concluded from spin-echo studies and from the observation of the linear electric field effect (LEFE) in spin-echo detected EPR that ‘The results indicate that the symmetry is not tetrahedral and that the ligation pattern is unlike that for either type 1 or type 2 Cu in other Cu proteins’ [13]. The Chan group applied continuous saturation to measure the electron spin relaxation parameter T l T 2 between 10 and 50 K for a variety of S = 1/2 species including type 1 and type 2 Cu centers, CuA of COX, inorganic Cu(II) complexes, and sulfur radicals. Over the entire temperature range examined, the relaxation of the type 1 coppers was much faster than that of type 2 copper, inorganic copper, and sulfur radicals. The relaxation of CuA of COX exhibited an unusual temperature dependence relative to the other copper complexes studied, suggesting a protein environment of this Cu site different from that of the other copper centers examined, and/or that CuA is influenced by a magnetic dipolar interaction with another, faster-relaxing paramagnetic site in the enzyme [14].
In this contribution, I would like to present an overview of the results of research we have performed in my laboratory for over three decades, in a fruitful collaboration with two great scientists and colleagues, Walter Zumft, head of the Institute of Molecular Microbiology (University of Karlsruhe), and William (Bill) Antholine from the National Biomedical EPR Center (Medical College of Wisconsin). Our collaborative work involved the application of a large variety of biochemical and biophysical methods to unravel the CuA active site present in COX and N2OR. Clearly, the discussion will be narrowly focused to some extent, yet hopefully it will help to illustrate what one may learn about studying the structure and function of a long-embattled, biological electron transfer center. This effort would not have been as successful without the input by outstanding researchers and their talented co-workers (cited in the references) with whom I had the privilege to interact, among them Helmut Beinert (University of Wisconsin), Gerhard Buse (RWTH Aachen), David Dooley (University of Rhode Island), Jürgen Hüttermann (Universität des Saarlandes), Jack Peisach (Albert Einstein College of Medicine), Matti Saraste (EMBL Heidelberg), Robert G. Scott (University of Georgia), Hans Thomann (ExxonMobil), Andrew Thomson (University of East Anglia), and William Tolman (University of Minnesota). Broader perspectives and more inclusive of the work of others in this dynamic research area are available in comprehensive reviews listed here [15,16,17,18,19].
A backward glance—early Anorganische Biochemie at Universities of Basel and Konstanz
I started my undergraduate studies at the University of Basel in 1962, where I met Helmut Sigel for the first time at the Institute of Inorganic Chemistry. Hans Erlenmeyer was the director of the institute, and he was not an inorganic chemist in the true sense of the word. He was fascinated by Warburg’s work on the role of metal ions in living matter [20]. Thus, not too surprising, Helmut Sigel did his Ph.D. thesis under the directorship of Prof. Erlenmeyer in an area of research nowadays called Bio-inspired Chemistry, or Inorganic Biological Chemistry [21, 22]. In 1968, I joined the group of Peter Hemmerich, another graduate student of Erlenmeyer, at the newly founded University of Konstanz to study Cu(I) complexes with nitrogen and sulfur ligands of biological interest. My Ph.D. supervisor, internationally best known for his pioneering achievements in the field of flavin chemistry [23, 24], was also keenly interested in the structure and function of catalytic groups in enzymes operating in conjunction with flavins such as the iron sulfur clusters or molybdenum, and in the binding of univalent and redox-active copper in proteins, especially the nature of copper in COX investigated by EPR spectroscopy. He strongly suggested to biochemists to consider sulfur ligands to copper in copper proteins and pointed out the resulting ambiguity in the valency of copper in such systems [25]. Hemmerich collaborated with Helmut Beinert from the Institute for Enzyme Research in Madison, and as early as 1966, purple, dinuclear copper mercaptide complexes of the type [Cu2(RS)2]2+ were discussed, formed from either Cu2+/RS− or Cu1+/RSSR (Eq. 1) [26], as well as binuclear mixed-valence copper complexes as models for Cu–Cu interaction in enzymes [27,28,29,30,31,32]. For further discussions of the sulfur non-innocence and the thiolate/disulfide-Cu(II)/Cu(I) redox couple (Eq. 1), more recent publications on this topic might be considered [33,34,35,36].
Early attempts to obtain a crystal structure of a dinuclear copper mercaptide, [Cu2(RS)2]2+ [R = –CH2CH(NH2)CO2 −, –CH2CH2NH2] remained without success [30], 5 years later, the X-ray structure of the purple \(\left[ {{\text{Cu}}^{\text{I}}_{8} {\text{Cu}}^{\text{II}}_{6} ({\text{RS}})_{12} {\text{Cl}}} \right]^{ 5- }\) cluster, [R = –C(Me2)CH(NH2)CO2 −], was reported, with eight S3-coordinated CuI atoms surrounding a central chloride ion [37]. Each CuII atom was N2S2– coordinated by two chelating RS− ligands. It took another two decades until the structural characterization of the first example of a thiolate-bridged, fully delocalized mixed-valence dicopper(I,II) complex was achieved by Bill Tolman and co-workers that modelled the CuA biological electron transfer site in COX and N2OR (discussed in section bio-inspired copper A complexes) [38, 39].
Two different Cu sites in cytochrome c oxidase—CuA and CuB
The history of understanding of the CuA site in COX from the early beginnings, when few believed that there was any significant Cu in COX (according to early studies by Warburg, the prosthetic group of COX, the Sauerstoffübertragende Ferment der Atmung, was a heme) [40, 41], to the verification of three Cu atoms/COX monomer and to the final identification of the CuA site as a dinuclear, Cys-bridged mixed-valence [Cu1.5+(CyS)2Cu1.5+] center through spectroscopy, recombinant DNA techniques, and X-ray crystallography was summarized in great detail by COX pioneer Beinert in two recent reviews [15, 16]. In his recollections, the critical advancements in understanding the nature of CuA are traced, and the contributions made by the development of methodology and concepts for solving the enigma of Cu in COX, are emphasized. Furthermore, the impediments, often rooted in contemporary preconceptions and attitudes rather than solid data, are discussed, which discouraged the exploitation of early valuable clues. At the very beginning, it was not known that there was more than one Cu site in COX. Notably, the presence of a second copper center, CuB, turned out to be much less controversial than the original notion that COX contained any functional Cu at all [42,43,44]. Later, from 1960 on, chemical analyses as well as numerous biochemical and EPR investigations had shown that there must be at least two significant Cu ions/two heme iron atoms in COX, and that one heme (a 3) as well as one Cu atom was not detectable by EPR [16]. The total Cu determined by chemical analysis could be accounted for by EPR, when the protein was denatured, and a portion of the second heme became EPR detectable upon partial reduction [7]. Although several explanations for these findings were considered, the suggestion that CuB and heme a 3 were closely linked so that the EPR signal was quenched by magnetic or electrostatic coupling appeared most plausible and has since been verified by other approaches including crystallography. The CuA site, however, resisted definitive identification for almost 40 years, close to the days when the X-ray structures of several COX proteins had been published [45,46,47,48,49]. As pointed out by Beinert, some of the best sources of detailed information on the early developments was often found in symposium contributions and review articles, which did allow space for lengthy discussions and most informative rebuttals, e.g., the symposium on copper in biological systems, held in 1965 at Arden House, New York [25], or the Manziana conferences on copper proteins [50,51,52], and the international meeting on the bioinorganic chemistry of copper, held in 1992 at Johns Hopkins University [53]. Needless to say, many of the conclusions presented in these references are now outdated and superseded by more advanced knowledge, however, these records document the many times painful progress of the problem’s maturation [16].
An important step forward resulted from the discovery of the absorption band centered around 830 nm. It turned out to be an optical marker for CuA in COX, as shown unambiguously by Thomson and co-workers. By elucidating the main magnetic circular dichroism (MCD) features at 475, 525 and 830 nm, they ascertained that the underlying chromophore had a spin of 1/2 and an EPR signal at g = 2 [54,55,56]. The availability of an optical marker for Cu at 830 nm helped to determine the capacity for electron uptake by COX. The reductive titrations, monitored by UV/Vis or EPR spectroscopy, suggested that there were two Cu2+ ions and two heme molecules in one monomer of oxidized COX. Remember that into the determination of such a stoichiometry several numbers are entering, of which each has a certain error attached to it. Consequently, it was difficult to discriminate between a stoichiometry of two heme: two Cu and one of two heme: three Cu. To demonstrate the difficulty, Chan et al. reported an extensive and accurate analyses of the Cu, Fe, Zn, and Mg contents in bovine heart COX, with the precision of an individual measurement within 25%, and a stoichiometry of 5Cu/4Fe/2Zn/2Mg per COX dimer [57]. Their preparations showed an “extra Cu” (Cux) which was removable by either monomerization of the enzyme or subunit III depletion, and it was argued that Cux played a structural role in enzyme dimerization. Decisive progress was achieved by Buse and co-workers through sequence analyses of the subunits of COX and in parallel by metal quantitation, from which it became apparent that there were actually three Cu atoms/two heme molecules in COX [58]. These authors did not just determine Fe and Cu but also Zn, Mg, and S by inductively coupled plasma atomic emission spectroscopy, so that the correlation of all these values from individual samples provided information on the homogeneity and purity of the protein. In addition, a stretch of amino acid sequence was identified in subunit II, which revealed a distinct similarity to the blue type 1 Cu sites, with two His, one Cys and one Met conserved at proper spacing. This led the authors to propose a binuclear copper site with a single cysteine bridge in COX subunit II (Fig. 2).

Tentative model of a binuclear copper site in subunit II of COX [58]
The structure of the CuA center is homologous to the cupredoxin fold that includes both the blue type 1 Cu proteins and the CuA electron transfer center [59,60,61,62]. Circular dichroism spectra of the CuA domain in COX documented the presence of the cupredoxin fold, a Greek key β-barrel and common structural motif in small, type1 Cu proteins of bacteria and plants [48]. This structural relationship was crucial to engineer the CuA dinuclear center into the type 1 Cu proteins amicyanin [63] and azurin by loop-directed mutagenesis [64, 65]. The observation of a sequence motif shared between the CuA center of N2OR and the subunit II of COX [66] led to the issue of a putative evolutionary relationship of the two enzymes [67]. The hypothesis of a common evolutionary origin of O2 and N-oxide respiratory enzymes was formulated explicitly on recognizing that NO reductase has structurally conserved metal-binding centers and extended sequence homology with the heme–Cu-type oxidase family [68, 69].
Nitrous oxide reductase, a multi-copper enzyme carrying a CuA site
The CuA center in COX, despite continuous efforts by many research groups applying a variety of different biochemical and biophysical techniques, resisted definitive identification for a long time. During this period of intense research, a crucial discovery was made in an apparently unrelated field, which soon had a decisive impact on the solution of the CuA problem. Walter Zumft at the University of Karlsruhe had identified the enzyme nitrous oxide reductase (N2OR) of denitrifying Pseudomonas stutzeri to be a novel copper protein [70, 71]. He was aware of my interest in copper proteins and the EPR facilities at the University of Konstanz, and in 1984, we received a first purple sample of N2OR to study its optical and EPR properties. This turned out to be the start of an extremely fruitful and successful collaboration for over 20 years [17], and culminated in the solution of the X-ray structure of the multi-copper enzyme by Anja Pomowski and Oliver Einsle at the University of Freiburg [72, 73]. The purple N2OR, isolated and purified under the strict exclusion of dioxygen (N2OR I), displayed an UV/Vis spectrum never reported before for a native copper protein, with an intense absorption maximum around 540 nm and a broadband centered around 800 nm (Fig. 3). Notably, the dithionite-reduced enzyme (N2OR III) was not colorless as expected for a diamagnetic Cu(I) 3d10 system but instead revealed a maximum around 650 nm and a rather featureless EPR signal with g values at 2.14 and 2.05. This signal recorded at X-band was assigned to the catalytic site for N2O reduction, called CuZ, somewhat analogous to the dioxygen reducing site Cub–Fe a3, in COX. It was later identified to be a novel tetranuclear copper site with coordinated inorganic sulfur [72,73,74,75,76,77,78,79].

UV/Vis (left) and EPR spectra (right) of the different forms of N2OR from Pseudomonas stutzeri. N2OR I, enzyme as isolated in the absence of dioxygen; N2OR III, N2OR I reduced with dithionite; N2OR IV, apo N2OR reconstituted with Cu(II)(en)2SO4; N2OR V, catalytically inactive derivative, isolated from mutant MK 402 [80, 81]
Even more interesting than the electronic spectrum was the EPR spectrum of the as-isolated N2OR (N2OR I) recorded at X-band (≈ 9.5 GHz), showing an unusual 7-line hyperfine pattern in the gII region (Fig. 3). Notably, this highly resolved 7-line EPR signal was only seen below 20 K, another unusual property for a Cu center as discussed below. In retrospect, a true example for the key message by Fred Hagen in his article EPR spectroscopy as a probe of metal centres in biological systems: in bio EPR spectroscopy, nothing compares to the returns from a standard X-band instrument [82]. The apparent hyperfine coupling A ║ vs g ║ (38 G, 120 MHz) was approximately 60% of the value expected for the blue type 1 Cu [83].
Choosing the right EPR frequency and temperature
In 1979, Froncisz and colleagues had reported a previously unseen hfs associated with the EPR-detectable Cu signal of COX, using low frequency 2–4 GHz EPR at 10 K. The hfs was consistent with hyperfine coupling to Cu, although to account for all of the observed structure, an additional magnetic interaction was required. The observed loss of resolution above 40 K had no parallel in the EPR literature of copper to effects seen from monomeric Cu complexes, suggesting an interaction with one of the other paramagnetic centers in COX, possibly heme a [84]. It is of interest to make reference here to a discussion by Beinert et al. in a footnote of their 1962 paper, mentioning the possibility of an exchange coupling between a Cu2+–Cu1+ pair in COX which (1) would account for the experimentally found 50% reduction for the integrated signal intensity, and (2) with the additional assumption of weak exchange coupling, would predict a hyperfine splitting constant, one-half of that which would be obtained from a Cu2+ ion alone [7]. However, in those days, the Cu:heme ratio in COX was believed to be 2:2, and not 3:2 as shown later by Buse et al. [58], and the multi-copper enzyme N2OR from denitrifying microorganisms had not yet been identified [17]. Obviously, the physical and biochemical properties of N2OR did not fit in the ‘classical’ scheme of type 1, 2 and 3 Cu proposed earlier [85], and in view of the broad absorption maximum around 800 nm in oxidized N2OR together with the position of A ║ vs g ║ in the Peisach–Blumberg diagram, these findings were a first indication that the EPR-detectable Cu in N2OR might have a structural and electronic arrangement similar to CuA of beef heart COX. Consequently, Bill Antholine and I initiated in 1987 the first Froncisz–Hyde experiments at the EPR Center in Milwaukee on concentrated samples of N2OR provided by Walter Zumft, and of COX provided by Gerhard Buse, using the loop gap resonator at frequencies in the range 2.5–5 GHz, at 20 K [86,87,88].
In parallel to these early EPR investigations at low microwave frequency, samples of N2OR I and of N2OR V were subjected to LEFE experiments in the laboratory of Jack Peisach (Albert Einstein College of Medicine) by his associate John McCracken. The LEFE effect is regarded to be a sensitive probe for mixed-valence Cu dimers and is expected to increase with increasing localization of the unpaired electron. The linear electric field effect found for CuA in N2OR was smaller than measured for type 1 centers [92]. These data were compared to LEFE data reported for different mixed-valence forms of the type 3 copper protein hemocyanin and type 1 Cu proteins [89], and supported the idea of a highly delocalized CuA ground state in N2OR. According to Mims, and Gerstman and Brill, a non-zero LEFE effect should only occur for molecules without a center of inversion [90, 91]. However, as in the case of parity forbidden electronic transitions, the LEFE can also be induced by vibronic coupling mechanisms; thus, the LEFE observed for CuA in N2OR may be attributed to vibronic coupling as well as a slight deviation from ideal symmetry (McCracken J, Neese F, Zumft WG, Kroneck PMH, Peisach J, unpublished data).
Based on the results obtained from comparative multifrequency EPR spectroscopy (2.5–35 GHz) of N2OR and COX, g-factor analysis, and computer simulation of the EPR spectra, a binuclear mixed-valence S = 1/2 site, [Cu1.5+–Cu1.5+], consisting of a 2Cu1.5+2SCys,2NHis core was proposed, as opposed to the ‘classical’ mononuclear site found in type 1 Cu proteins, i.e. a Cu2+SCys,2 NHis,SMet center [93, 94] (Fig. 4).

Structural model for the CuA rhomb in N2OR from Pseudomonas stutzeri derived from spectroscopic and mutagenesis studies, and analogy to the reported CuA crystal structures [92]
This model of a mixed-valent CuA rhomb in N2OR and COX was refined as discussed below, and later confirmed by high-resolution X-ray crystallography, however, with a surprising result [72, 73]. In N2OR, in contrast to COX, the CuA center appeared to be structurally flexible. His 583 was not a ligand to CuA in the enzyme as isolated under exclusion of dioxygen, but rotated by 130° to form a hydrogen bond to residue Ser 550. This led to a shortened Cu–Cu distance and thus likely to a sharpening of the characteristic 7-line hyperfine pattern. Upon binding of the substrate N2O to the catalytic CuZ center, the ligands of CuA rearranged and His 583 moved to ligate to CuA, opening an efficient electron transfer pathway from Asp 576, a presumed entry point for electrons delivered by the physiological redox partner of the enzyme (Fig. 5). Earlier, using a binuclear CuA site masterly engineered into the type 1 copper protein azurin by Yi Lu and co-workers [95,96,97] and mutagenesis of the equatorial His 120 ligand, multifrequency EPR and ENDOR experiments were carried out to understand the histidine-related modulation to CuA, notably to the valence delocalization over the [Cu1.5+–Cu1.5+] core. According to the ENDOR results, the CuA core electronic structure remained unchanged by the His 120 mutation. On the other hand, multifrequency EPR indicated that the H120N and H120G mutations had changed the EPR hyperfine signature from a 7-line to a 4-line pattern, consistent with a trapped-valence, type 1 mononuclear copper [98]. Along these lines, Zickermann et al. produced another interesting CuA variant, by replacing the distant Met 227 ligand of Paracoccus denitrificans COX with isoleucine. The EPR spectrum and near-infrared absorption were changed by this substitution, however, the Cu content and the dimeric structure were maintained. The variant still exhibited the mixed-valence feature but the Cu atoms were no longer equivalent resulting in a localized [Cu2+–Cu1+] CuA site, and the e-transfer rate between CuA and heme a was diminished [99].

CuA site of N2OR from Pseudomonas stutzeri. a Stereo representation of CuA as isolated under the exclusion of dioxygen. Contrary to all structures available for the conserved CuA center, His 583 is not a ligand to a copper ion. b Upon binding of N2O to catalytic center CuZ, His 583 flips to ligate to CuA. PDB ID 3SBR [72, 73]
The suggestion of a binuclear copper site for CuA in COX based on the EPR spectroscopic properties of N2OR generated quite some controversy [100, 101]. Bo Malmström, a leading researcher and pioneer in the field of copper proteins, pointed out that the presence of a third functional copper in COX, as suggested by Buse and co-workers [58], was difficult to accept in view of the complete lack of an extra Cu EPR signal. Ascribing the CuA EPR spectrum to a binuclear Cu site, with one unpaired electron delocalized between two equivalent Cu nuclei, was a clever solution to this dilemma, unfortunately, this proposal was undoubtedly wrong according to Malmström [102]. The issue of mononuclear vs binuclear CuA in COX became latest settled with the crystallographic structure of COX [45, 46, 103], and of the membrane-exposed domain from a respiratory quinol oxidase complex with an engineered binuclear copper center (Fig. 6) [104].

Clearly, the X-band EPR spectra of bacterial and mammalian COX, as well as of engineered soluble CuA fragments, did not show the well-resolved 7-line pattern observed for N2OR I at g II = 2.18 [17, and discussion herein]. The second harmonic display, or pseudomodulation treatment of the EPR spectra of COX helped to achieve some resolution enhancement (Fig. 7) [94].

Left: X-band EPR spectra (second harmonic display) of a 65Cu-enriched and b 65Cu,15N-histidine-enriched N2OR from Pseudomonas stutzeri. Temperature 10 K; microwave power 200 µW; microwave frequency 9.24128 GHz; modulation amplitude 4G. Right: Simulated second derivative spectra of CuA in N2OR. a Experimental spectrum of 15N-histidine/65Cu N2OR b Simulated spectrum using non-collinear g and ACu tensors c Simulated spectrum using collinear g and ACu tensors [92, 106]
In my view, it is fair to say that it was Frank Neese, a brilliant Ph.D. student in my group, who developed the first in-depth picture of the electronic structure of CuA [92, 105]. The EPR spectra of either 63Cu or 65Cu-enriched N2OR firmly established the mixed-valence delocalized configuration. The EPR spectrum after insertion of both 65Cu and 15N-histidine improved the resolution of the Cu hyperfine lines, but no superhyperfine lines from nitrogen and proton coupling were resolved (Fig. 7).
To refine the simulation of the EPR spectra mixed-valence theory was applied. The [Cu1.5+–Cu1.5+] center was treated as a dimer built from two interacting monomeric sites CuA1 and CuA2. The values obtained were similar to those reported earlier by [94]. The refinement in the simulation was achieved by treating the two Cu ions to be slightly inequivalent and the introduction of the non-collinearity between the system-g and metal hyperfine tensors (Figs. 7, 8).

Schematic representation of the relationship between the g and A main axes and construction of the g-tensor in a delocalized mixed-valence [Cu1.5+–Cu1.5+] site from hypothetical subsite g tensors of CuA1 and CuA2. The inset shows the predicted behavior of the principle values of the g-tensor as a function of local magnetic axes tilt [92, 106]
In consecutive steps, the electronic picture of CuA in both N2OR and COX was refined by F. Neese in collaboration with A. Thomson, M. Saraste, J. Hüttermann, and their associates. Based on X-ray structural data of the engineered CuA center (Fig. 6) [104], molecular orbital calculations on a sulfur-bridged [(NH3)Cu1.5+(SCH3)2Cu1.5+(NH3)]+ core were used to interpret the EPR spectroscopic results. The calculated spin distribution showed an excellent agreement with the values derived from the analysis of the Cu hyperfine structure [106, 107]. ENDOR spectra for the [Cu1.5+–Cu1.5+], S = 1/2 site were obtained of the 63Cu, 65Cu, or 65Cu, 15N-histidine enriched enzyme [108]. The 14N/15N isotopic substitution allowed for an unambiguous deconvolution of proton and nitrogen hyperfine structure in the spectra. The anisotropy in the nitrogen hyperfine values was characteristic for imidazole bound to copper, and the [(NH3)Cu1.5+(SCH3)2Cu1.5+(NH3)]+ core structure was predicted to be similar to the CuA structure determined by Wilmanns et al. (Fig. 6) [104]. In the CuA center, the unpaired spin is almost equally distributed over the Cu and S atoms in the electronic ground state, and there is only 3–5% spin density on the nitrogen donors of CuA. Consequently, CuA in its electronic ground state is viewed best as a completely delocalized radical in which the delocalization occurs via the in-plane σ/π framework rather than via the out-of-plane π framework as for delocalized organic radicals. This situation may be described by the resonance structures [CuIIRS−RS−CuI \(\leftrightarrow\) CuIRS•RS−CuI \(\leftrightarrow\) CuIRS−RS•CuI \(\leftrightarrow\) CuIRS−RS−CuII] [92]. The theoretical calculations carried out by Neese, Thomson, and co-workers to develop a molecular orbital picture of the binuclear mixed-valent center, were followed by similar calculations from other groups. To get a deeper insight, electronic structural descriptions were developed for several CuA-type centers and structurally characterized copper thiolates. Both the Cu–Cu compression and removal of the axial ligands appeared to be critical determinants of the orbital ground state in these dimeric systems [109,110,111,112].
Fast relaxation, 1H NMR spectroscopy, and copper–copper bond in CuA
A characteristic physical property of the CuA site in both N2OR and COX is its extremely fast relaxation. The product T 1 T 2 of CuA in COX exhibited a stronger temperature dependence than any of the type 1 or type 2 Cu(II) sites, and it was proposed that CuA was influenced by a magnetic dipolar interaction with another, faster-relaxing paramagnetic site [14, 84]. In collaboration with Hassane Mcharaourab and Susanne Pfenninger, the multiquantum EPR technique was applied, and pure absorption spectra for CuA were obtained [113]. Saturation recovery data confirmed that the intrinsic electron spin–lattice relaxation time, T 1, for N2OR in the range 6–25 K was unusually short for copper centers. The short T 1 was attributed to the vibrational modes of type-1 and/or the metal–metal interaction in [Cu1.5+–Cu1.5+] [114]. The unusual relaxation properties of the CuA center allowed extensive NMR investigations of CuA-containing protein fragments in solution [115,116,117,118,119,120], and of N2OR from P. stutzeri [121]. These experiments were crucial (1) to assign the NMR signals of CuA ligands and interaction with surrounding amino acid residues, (2) to advance possible mechanisms to understand the extremely fast electron relaxation of the CuA site and its flexibility, and (3) to provide further insights into the electronic structure of the CuA site at atomic resolution in solution at physiologically relevant temperature.
Another intriguing feature of the CuA rhomb is related to its short Cu–Cu distance, as deduced from a Cu K-edge EXAFS study of the soluble CuA fragment from COX of Bacillus subtilis which indicated a directly bonded Cu–Cu unit, with a Cu–Cu distance of approximately 2.5 Å like in metallic copper [122]. Each Cu atom was coordinated to one histidine and two cysteine residues, and a Cu–Cu metal bond was suggested. Based on calculations of the electronic structure and spectroscopic properties of the CuA center, the model structure consisted of a dimer of two type 1 Cu centers, with no bridging ligands but a direct Cu–Cu bond [123, 124]. The Cu K-edge X-ray absorption spectrum of bovine heart COX prepared in the laboratory of G. Buse also provided evidence for a binuclear Cu center [125]. Cu K-edge EXAFS studies on mixed-valent [Cu1.5+–Cu1.5+] and reduced [Cu1+–Cu1+] species refined the structure of the Cu2S2 rhomb (also called diamond), yielding a Cu–Cu distance of 2.43 Å for the mixed-valence, and 2.51 Å for the one electron reduced center in CuA fragments of COX from B. subtilis and Thermus thermophilus. The short Cu–Cu distance could be the result, in part, of a weak metal–metal bond. It was suggested that the function of the CuA cluster was to provide a site with minimal structural perturbation occurring during electron transfer, thus, offering an excellent rationalization for the very low reorganizational energy observed for the CuA center [122].
Bio-inspired CuA complexes
I want to come back here to remarks by Hemmerich at a meeting on the biochemistry of copper in 1965 [25], where he discussed the manifold possibilities one could expect in copper–thiol interactions, and where he pointed out the possible existence of the [Cu(μ-RS)2Cu]+ core in COX and as a transient species in Cu thiolate systems [26]. Early attempts to model the spectroscopic properties by biomimetic CuS2N2 complexes [126] remained without success to produce model compounds with EPR, optical or MCD spectra similar to those of the CuA site of COX. Nelson and co-workers prepared a Cu(I), Cu(II) mixed-valence complex that was later structurally and spectroscopically characterized [127,128,129]. The complex [Cu2(LiPrdacoS)2]+ was the first example of a structurally characterized thiolate-bridged mixed-valence copper dimer, specifically designed to mimic the physical properties of the CuA site [38, 39]. More recently, a series of fully delocalized [Cu1.5+–Cu1.5+] mixed-valence complexes has been assembled, having only oxygen donor bonds [130], a 1,8-naphthyridine-based dinucleating ligand [131], two ureate bridging ligands [132], the novel Cu2N2 diamond core [133], or (1,1,3,3-tetramethylguanidine) benzenethiolate [134].
Outlook and concluding remarks
The redox enzymes N2OR and COX are constituents of important biological processes. N2OR is the terminal reductase in a respiratory chain converting N2O to N2 in denitrifying bacteria, COX is the terminal oxidase of the aerobic respiratory chain of certain bacteria and eukaryotic organisms. Both enzymes are involved in complex multi-electron and multi-proton transfer reactions of kinetically inert gaseous molecules (Eqs. 2, 3). Notably, COX also acts as a proton pump [103, 135,136,137,138,139].
A wide array of different spectroscopies was applied to show that bacterial N2OR had a copper site with properties very similar to the CuA site of COX [17]. Fortunately, as there were no heme chromophores in N2OR, its spectroscopic features could be analyzed without interference from heme. Characteristic Cu hfs was observed in the EPR spectra indicative for a mixed-valence Cu site. To exclude the interference by the tetranuclear CuZ catalytic center, Walter Zumft succeeded to produce a catalytically inactive N2OR (N2ORV) which lacked the catalytic CuZ but contained the electron transferring CuA. These preparations, labeled specifically with a single Cu isotope, allowed a careful study of the seven-line hyperfine pattern of CuA from N2OR at low (2.4–4.6 GHz) microwave frequencies. The 7-line pattern due to Cu, with an intensity ratio of 1:2:3:4:3:2:1 for individual lines, firmly established the existence of the mixed-valence [Cul.5+–Cu1.5+] pair in N2OR [17]. The proposal that COX also contained a binuclear mixed-valent copper site was initially opposed by leading researchers in the field, among them COX pioneers Sunney Chan, Bo Malmström, and not to forget Helmut Beinert. He once pointed out to me that CuA in COX had been the graveyard for many splendid ideas, and that we, Antholine, Kroneck, and Zumft, should be cautious to contribute not another one. However, his input was crucial to reply to the criticism brought forward by Chan and Malmström [100]. Helmut Beinert also helped to get a sample of COX from shark, which was known to be a monomer in the native state contrary to the enzyme from bovine heart [140]. To our relief, this sample also exhibited the characteristic multiline EPR spectrum found in dimeric COX [see discussion about Cux [57]; Antholine WE, Zumft WG, Kroneck PMH, Beinert H, unpublished results]. In this context, one should remember that the stoichiometry of three Cu atoms/COX monomer was not generally accepted. However, when it became clear that the amino acid sequences of N2OR and COX showed a pattern of two similarly spaced His, Cys and a Met residue, the proposal by Antholine, Kroneck, and Zumft [17] seemed to be the best interpretation of all analytical and spectroscopic data. In addition, the uptake of a maximum of four electrons by one COX monomer could also be understood. In other words, the CuA center shuttled between redox states [Cu1.5+–Cu1.5+]3+ and [Cu1+–Cu1+]2+, consuming one electron per two Cu, leaving three electrons for heme a and the dioxygen-reducing center hemea 3–CuB.
Once the mixed-valent, binuclear nature of CuA in COX and N2OR had gained general acceptance (see remarks about “When a thing is new…” by William James, known as the ‘nitrous oxide philosopher’ [17]), an impressive number of important contributions to our understanding of CuA has been published by prominent researchers in the field of copper biochemistry, among them Ivano Bertini (CERM, Florence), Ninian Blackburn (Oregon Health and Science University), Gerard Canters (Leiden University), Daniella Goldfarb (Weizmann Institute of Science), Ole Favre (University of Copenhagen), Jim Fee (The Scripps Research Institute), Yi Lu (University of Illinois), Israel Pecht (Weizmann Institute of Science), Edward (Ed) Solomon (Stanford University), and AlejandroVila (Instituto de Biología Molecular y Celular de Rosario), to name a few. In first place, in my view, comes the Stanford group under the lead of Ed Solomon. Thanks to the work by Ed and his associates, the major geometric and electronic features of the CuA rhomb, including metal–ligand covalency, Cu–Cu metal bond, redox potentials, reorganization energies, valence delocalization, and the weakened axial bonding interactions in relation to its electron transfer function, and specific potential electron transfer pathways can be analyzed at the sub-atomic level [19 and references therein]. In this context, I want to bring up the fourth commandment of A. Kornberg remembering the late Efraim Racker: “Do not waste clean thinking on dirty enzymes” [141]. Over many years, we received large quantities of high-quality enzyme samples from either Walter Zumft (bacterial N2OR), or from Gerhard Buse (beef heart COX), to perform advanced biochemical and biophysical experiments. In a similar way, the Stanford group had the privilege to collaborate with two outstanding scientists, on the one hand Yi Lu, the bioengineer of complex metal centers [18, 142], and on the other hand Bill Tolman, who promised to synthesize a mixed-valent Cu(I), Cu(II) mercaptide complex at a time, when the binuclear nature of CuA was still under debate [143, 144].
Admittedly, not all our experiments with high-quality N2OR were successful, such as the extensive pulse radiolysis studies on electron transfer reactions between CuA and the catalytic CuZ site together with Ole Farver and Israel Pecht at the Weizmann Institute of Science. It took an engineered purple CuA azurin to demonstrate that the binuclear purple CuA center was a more efficient electron transfer agent than the mononuclear blue type 1 Cu center because reactivity of the former involved lower reorganization energy [145]. Finally, the positioning of the substrate N2O observed in the X-ray structure of N2OR between the two metal sites, CuA and CuZ, points towards a reaction mechanism where the substrate, precisely oriented by the protein, becomes activated by the tetranuclear CuZ cluster. Only after binding of N2O is electron transfer from CuA to CuZ observed, consequently, rather than being a mere electron transfer site, CuA of N2OR appears to be an integral part of the enzyme’s catalytic center (Fig. 9).

The active site of Pseudomonas stutzeri N2OR with bound substrate N2O. The stereo image shows CuA of the cupredoxin subunit (blue) interacting closely with CuZ from the β-propeller subunit (green) in the N2OR head-to-tail dimer. N2O binds in a side-on orientation on the face of the CuZ cluster, oriented by conserved amino acid residues from the cupredoxin domain. This binding mode most likely allows for direct electron transfer from the CuA site to the substrate, making the complex arrangement of two metals sites plus the intervening protein residues from two subunits a single, intricate active centre for N2O reductase. PDB ID 3SBR [72, 73]
References
Sigel H, Brintzinger H (1963) Helv Chim Acta 46:701–712
Sigel H, Brintzinger HH, Erlenmeyer H (1963) Helv Chim Acta 46:712–719
Sigel H, Brintzinger HH (1964) Helv Chim Acta 47:1701–1717. https://doi.org/10.1002/hlca.19640470706
McCormick DB, Griesser R, Sigel H (1974) Met Ions Biol Syst 1:213–247
Sands RH, Beinert H (1959) Biochem Biophys Commun 1:175–178. https://doi.org/10.1016/0006-291X(59)90013-0
Beinert H, Griffiths DE, Wharton DC, Sands RH (1962) J Biol Chem 237:2337–2346
Peisach J, Blumberg WE (1974) Arch Biochem Biophys 165:691–708
Vänngård T (1972) In: Swartz HM, Bolton JR, Borg DC (eds) Copper proteins. Wiley, New York, pp 411–447
Stevens TH, Martin CT, Wang H, Brudvig GW, Scholes CP, Chan SI (1982) J Biol Chem 257:12106–12113
Chan SI, Li PM (1990) Biochemistry 29:1–12
Gurbiel RJ, Fann Y-C, Surerus KK, Werst MM, Musser SM, Doan PE, Chan SI, Fee JA, Hoffman BM (1993) J Am Chem Soc 115:10888–10894
Mims WB, Peisach J, Shaw RW, Beinert H (1980) J Biol Chem 255:6843–6846
Brudvig GW, Blair DF, Chan SI (1984) J Biol Chem 259:11001–11009
Beinert H (1995) Chem Biol 2:781–785
Beinert H (1997) Eur J Biochem 245:521–532
Zumft WG, Kroneck PMH (2006) Adv Microb Physiol 52(107–22):7. https://doi.org/10.1016/S0065-2911(06)52003-X
Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y (2014) Chem Rev 114:4366–4469
Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L (2014) Chem Rev 114:3659–3853. https://doi.org/10.1021/cr400327t
Bloch H (1969) Basler Stadtbuch, 37–40. https://www.baslerstadtbuch.ch/.permalink/stadtbuch/16fe260b-51f4-47d1-b298-ff3691d9d265
Valentine J, O’Halloran TV (1999) Curr Opin Chem Biol 3:129–130
Holm RH, Solomon EI (2004) Chem Rev 104:347–348
Hemmerich P, Veeger C, Wood HCS (1965) Angew Chem Int Ed 4:671–687
Beinert H, Massey V (1982) Trends Biochem Sci 7:43–44. https://doi.org/10.1016/0968-0004(82)90069-X
Hemmerich P (1966) In: Peisach J, Aisen P, Blumberg WE (eds) The biochemistry of copper. Academic Press, New York, pp 15–34
Hemmerich P, Beinert H, Vänngård T (1966) Angew Chem Int Ed 5:422–423
Hemmerich P, Sigwart C (1963) Experientia 19:488–489
Sigwart C, Hemmerich P, Spence JT (1968) Inorg Chem 7:2545–2548. https://doi.org/10.1021/ic50070a015
Sigwart C, Kroneck PMH, Hemmerich P (1970) Helv Chim Acta 53:177–185. https://doi.org/10.1002/hlca.19700530125
Kroneck PMH, Naumann C, Hemmerich P (1971) Inorg Nucl Chem Lett 7:659–666. https://doi.org/10.1016/0020-1650(71)80052-1
Kroneck PMH (1975) J Am Chem Soc 97:3839–3841. https://doi.org/10.1021/ja00846a059
Vortisch V, Kroneck PMH, Hemmerich P (1976) J Am Chem Soc 98:2821–2826. https://doi.org/10.1021/ja00426a025
Itoh S, Nagagawa M, Fukuzumi S (2001) J Am Chem Soc 123:4087–4088
Neuba A, Haase R, Meyer-Klaucke W, Flörke U, Henkel G (2012) Angew Chem Int Ed 51:1714–1718
Thomas AM, Lin B-L, Wasinger EC, Stack TDP (2013) J Am Chem Soc 135:18912–18919
Ording-Wenker ECM, van der Plas M, Siegler MA, Bonnet S, Bickelhaupt FM, Fonseca Guerra C, Bouwman E (2014) Inorg Chem 53:8494–8504. https://doi.org/10.1021/ic501060w
Birker PJML, Freeman HC (1976) J Chem Soc Chem Commun 9:312–313. https://doi.org/10.1039/C39760000312
Houser RP, Young VG, Tolman WB (1995) J Am Chem Soc 117:10745–10746. https://doi.org/10.1021/ja00148a018
Houser RP, Young VG, Tolman WB (1996) J Am Chem Soc 118:2101–2102. https://doi.org/10.1021/ja953776m
Warburg O (1932) Angew Chem 45:1–6. https://doi.org/10.1002/ange.19320450102
Person P, Wainio WW, Eichels B (1953) J Biol Chem 202:369–381
Elvehjem CA (1931) J Biol Chem 90:111–132
Cohen E, Elvehjem CA (1934) J Biol Chem 107:97–105
Keilin D, Hartree EF (1939) Proc R Soc (Lond.) B127:167–191
Iwata S, Ostermeier C, Ludwig B, Michel H (1995) Nature 37:660–669
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S (1995) Science 269:1069–1074
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa R (1996) Science 272:1136–1144
Williams PA, Blackburn NJ, Sanders D, Bellamy H, Stura EA, Fee JA, McRee DE (1999) Nat Struct Biol 6:509–516
Soulimane T, Buse G, Bourenkov GP, Bartunik H, Huber R, Than ME (2000) EMBO J 19:1766–1777
Beinert H (1980) Coord Chem Rev 33:55. https://doi.org/10.1016/S0010-8545(00)80398-7
Beinert H (1991) J Inorg Biochem 44:173–218. https://doi.org/10.1016/0162-0134(91)80054-L
Beinert H (1996) J Inorg Biochem 64:79–100. https://doi.org/10.1016/0162-0134(96)00083-9
Karlin KD, Tyeklár Z (1993) Bioinorganic chemistry of copper. Chapman & Hall, New York
Greenwood C, Hill BC, Barber D, Eglinton DG, Thomson AJ (1983) Biochem J 215:303–316
Thomson AJ, Greenwood C, Peterson J, Barrett CP (1986) J Inorg Biochem 28:195–205
Greenwood C, Thomson AJ, Barrett CP, Peterson J, George GN, Fee JA, Reichardt J (1988) Ann N Y Acad Sci 550:47–52
Pan LP, Li Z, Larsen R, Chan SI (1991) J Biol Chem 266:1367–1370
Steffens GCM, Soulimane T, Wolff G, Buse G (1993) Eur J Biochem 213:1149–1157
Adman ET (1991) Adv Protein Chem 42:145–197
Dennison C, Canters GW (1996) Recl Trav Chim Pays Bas 115:345–351
Vila AJ, Fernandez CO (2001) Copper in electron transfer proteins. In: Bertini I, Sigel A, Sigel H (eds) Handbook on metalloproteins. Marcel Dekker, New York, pp 813–856
Lu Y (2003) Cupredoxins. In: Que L, Tolman WB (eds) Biocoordination chemistry. Comprehensive coordination chemistry II: from biology to nanotechnology, vol 8. Elsevier, Oxford, pp 1–32
Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters GW (1995) FEBS Lett 365:92–94
Hay M, Richards JH, Lu Y (1996) Proc Natl Acad Sci USA 93:461–464
Robinson H, Ang MC, Gao Y-G, Hay MT, Lu Y, Wang AH-J (1999) Biochemistry 38:5677–5683
Viebrock A, Zumft WG (1988) J Bacteriol 170:4658–4668
Zumft WG (1992) The denitrifying prokaryotes. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes. A handbook on the biology of bacteria: ecophysiology, isolation, identification, applications, vol 1. Springer, New York, pp 554–582
Saraste M, Castresana J (1994) FEBS Lett 341:1–4
van der Oost J, de Boer APN, de Gier JWL, Zumft WG, Stouthamer AH, van Spanning RJM (1994) FEMS Microbiol Lett 121:1–10
Zumft WG, Matsubara T (1982) FEBS Lett 148:102–107
Zumft WG, Coyle CL, Frunzke K (1985) FEBS Lett 183:240–244
Pomowski A, Zumft WG, Kroneck PMH, Einsle O (2011) Nature 477:234–238. https://doi.org/10.1038/nature10332
Wüst A, Schneider L, Pomowski A, Zumft WG, Kroneck PMH, Einsle O (2012) Biol Chem 393:1067–1077. https://doi.org/10.1515/hsz-2012-01771
Brown K, Tegoni M, Prudêncio M, Pereira AS, Besson S, Moura JJG, Moura I, Cambillau C (2000) Nat Struct Biol 7:191–195
Rasmussen T, Berks BC, Sanders-Loehr J, Dooley DM, Zumft WG, Thomson AJ (2000) Biochemistry 39:12753–12756
Brown K, Djinovic-Carugo K, Haltia T, Cabrito I, Saraste M, Moura JJG, Moura I, Tegoni M, Cambillau C (2000) J Biol Chem 275:41133–41136
Rasmussen T, Berks BC, Butt JN, Thomson AJ (2002) Biochem J 364:807–815
Haltia T, Brown K, Tegoni M, Cambillau C, Saraste M, Mattila K, Djinovic-Carugo K (2003) Biochem J 369:77–88
Oganesyan VS, Rasmussen T, Fairhurst S, Thomson AJ (2004) Dalton Trans 7:996–1002
Coyle CL, Zumft WG, Kroneck PMH, Körner H, Jakob W (1985) Eur J Biochem 153:459–467. https://doi.org/10.1111/j.1432-1033.1985.tb09324.x
Riester J, Zumft WG, Kroneck PMH (1989) Eur J Biochem 178:751–762. https://doi.org/10.1111/j.1432-1033.1989.tb14506.x
Hagen WR (2006) Dalton Trans 37:4415–4434. https://doi.org/10.1039/B608163K
Solomon EI, Lowery MD (1993) Science 259:1575–1581
Froncisz W, Scholes CP, Hyde JS, Wei Y-H, King TE, Shaw RW, Beinert H (1979) J Biol Chem 254:7482–7484
Malkin R, Malmström BG (1970) Adv Enzymol 33:177–244
Froncisz W, Hyde JS (1980) J Chem Phys 73:3123–3131
Froncisz W, Hyde JS (1982) J Magn Reson 47:515–521
Kroneck PMH, Antholine WE, Riester J, Zumft WG (1988) FEBS Lett 242:70–74
Westmoreland TD, Wilcox DE, Baldwin MJ, Mims WB, Solomon EI (1989) J Am Chem Soc 111:6106–6123
Mims WB (1976) The linear electric field effect in paramagnetic resonance. Clarendon Press, Oxford
Gerstman BS, Brill AS (1988) Phys Rev A 37:2151–2164
Neese F (1997) Electronic structure and spectroscopy of novel copper chromophores in biology. Ph.D. Dissertation, University of Konstanz, Germany
Kroneck PMH, Antholine WE, Kastrau DHW, Buse G, Steffens GCM, Zumft WG (1990) FEBS Lett 268:274–276. https://doi.org/10.1016/0014-5793(90)81026-K
Antholine WE, Kastrau DHW, Steffens GCM, Buse G, Zumft WG, Kroneck PMH (1992) Eur J Biochem 209:875–881. https://doi.org/10.1111/j.1432-1033.1992.tb17360.x
Hay MT, Lu Y (2000) J Biol Inorg Chem 5:699–712
Savelieff MG, Wilson TD, Elias Y, Nilges MJ, Garner DK, Lu Y (2008) Proc Natl Acad Sci (USA) 105:7919–7924
Savelieff M, Lu Y (2010) J Biol Inorg Chem 15:461–483
Lukoyanov D, Berry SM, Lu Y, Antholine WE, Scholes CP (2002) Biophys J 82:2758–2766
Zickermann V, Verkhovsky M, Morgan J, Wikström M, Anemüller S, Bill E, Steffens GCM, Ludwig B (1995) Eur J Biochem 234:686–693
Li PM, Malmström BG, Chan SI (1989) FEBS Lett 248:210–211
Kroneck PMH, Antholine WE, Riester J, Zumft WG (1989) FEBS Lett 248:212–213
Malmström BG (1990) Arch Biochem Biophys 280:233–241
Yoshikawa S, Shimada A, Shinzawa-Itoh K (2015) Met Ions Life Sci 15:89–130
Wilmanns M, Lappalainen P, Kelly M, Sauer-Eriksson E, Saraste M (1995) Proc Natl Acad Sci USA 92:11955–11959
Sinnecker S, Neese F (2007) Top Curr Chem 268:47–83. https://doi.org/10.1007/128_2006_081
Neese F, Zumft WG, Antholine WE, Kroneck PMH (1996) J Am Chem Soc 118:8692–8699
Farrar JA, Neese F, Lappalainen P, Kroneck PMH, Saraste M, Zumft WG, Thomson AJ (1996) J Am Chem Soc 118:11501–11514
Neese F, Kappl R, Hüttermann J, Zumft WG, Kroneck PMH (1998) J Biol Inorg Chem 3:53–67
Gamelin DR, Randall DW, Hay MT, Houser RP, Mulder TC, Canters GW, de Vries S, Tolman WB, Lu Y, Solomon EI (1998) J Am Chem Soc 120:5246–5526
Randall GW, Gamelin DR, LaCroix LB, Solomon EI (2000) J Biol Inorg Chem 5:16–19
DeBeer George S, Metz M, Szilagyi RK, Wang H, Cramer SP, Lu Y, Tolman WB, Hedman B, Hodgson KO, Solomon EI (2001) J Am Chem Soc 123:5757–5767
Olsson MHM, Ryde U (2001) J Am Chem Soc 123:7866–7876
Mchaourab HS, Pfenninger S, Antholine WE, Felix CC, Hyde JS, Kroneck PMH (1993) Biophys J 64:1576–1579
Pfenninger S, Antholine WE, Barr ME, Hyde JS, Kroneck PMH, Zumft WG (1995) Biophys J 69:2761–2769
Bertini I, Bren KL, Clemente A, Fee JA, Gray HB, Luchinat C, Malmström BG, Richards JH, Sanders D, Slutter CE (1996) J Am Chem Soc 118:11658–11659
Luchinat C, Soriano A, Djinovic-Carugo K, Saraste M, Malmström BG, Bertini I (1997) J Am Chem Soc 119:11023–11027
Salgado J, Warmerdam G, Bubacco L, Canters GW (1998) Biochemistry 19:7378–7389
Abriata LA, Ledesma GN, Pierattelli R, Vila AJ (2009) J Am Chem Soc 131:1939–1946
Zaballa M-E, Luciano LA, Donaire A, Vila AJ (2012) Proc Natl Acad Sci USA 109:9254–9259
Alvarez-Paggi D, Zitare UA, Szuster J, Morgada MN, Leguto AJ, Vila AJ, Murgida DH (2017) J Am Chem Soc 139:9803–9806
Holz RC, Alvarez ML, Zumft WG, Dooley DM (1999) Biochemistry 38:11164–11171
Blackburn NJ, de Vries S, Barr ME, Houser RP, Tolman WB, Sanders D, Fee JA (1997) J Am Chem Soc 119:6135–6143
Larsson S, Källebring B, Wittung P, Malmström BG (1995) Proc Natl Acad Sci USA 92:7167–7171
Ramirez BE, Malmström BG, Winkler JR, Gray HB (1995) Proc Natl Acad Sci USA 92:11949–11951
Henkel G, Müller A, Weissgräber S, Buse G, Soulimane T, Steffens GCM, Nolting H-F (1995) Angew Chem Int Ed 34:1488–1492
Toftlund H, Becher J, Olesen PH, Pedersen JZ (1985) Isr J Chem 25:56–65
Harding C, McKee V, Nelson J (1991) J Am Chem Soc 113:9684–9685. https://doi.org/10.1021/ja00025a050
Farrar JA, McKee V, Al-Obaidi AHR, McGarvey JJ, Nelson J, Thomson AJ (1995) Inorg Chem 34:1302–1303
Kababya S, Nelson J, Calle C, Neese F, Goldfarb D (2006) J Am Chem Soc 128:2017–2029. https://doi.org/10.1021/ja056207f
LeCloux DD, Davydov R, Lippard SJ (1998) Inorg Chem 37:6814–6826
He C, Lippard SJ (2000) Inorg Chem 39:5225–5231
Gupta R, Zhang ZH, Powell D, Hendrich MP, Borovik AS (2002) Inorg Chem 41:5100–5106
Harkins SB, Peters JC (2004) J Am Chem Soc 126:2885–2893
Witte M, Grimm-Lebsanft B, Goos A, Binder S, Rübhausen M, Bernard M, Neuba A, Gorelsky S, Gerstmann U, Henkel G, Schmidt WG, Herres-Pawlis S (2016) J Comp Chem 37:2181–2192. https://doi.org/10.1002/jcc.24439
Zumft WG (1997) Microbiol Mol Biol Rev 61:533–616
Schneider LK, Wüst A, Pomowski A, Zhang L, Einsle O (2014) Met Ions Life Sci 14:177–210
Michel H (1998) Proc Natl Acad Sci USA 95:12819–12824
Belevich I, Verkhovsky MI, Wikström M (2006) Nature 440:829–832
Yoshikawa S, Muramoto K, Shinzawa-Itoh K (2011) Annu Rev Biophys 40:205–223
Wilson MT, Lalla-Maharajh W, Darley-Usmar V, Bonaventura J, Bonaventura C, Brunori M (1980) J Biol Chem 255:2722–2728
Kornberg A (2000) J Bacteriol 182:3613–3618
Lu Y (2006) Angew Chem Int Ed 45:5588–5601
Tolman WB, Carrier SM, Ruggiero CE, Antholine WE, Whittacker J (1993) In: Karlin KD, Tyeklár Z (eds) Bioinorganic chemistry of copper. Chapman & Hall, New York, pp 406–418
Tolman WB (2010) Angew Chem Int Ed 49:1018–1024
Farver O, Lu Y, Ang MC, Pecht I (1999) Proc Natl Acad Sci USA 96:899–902
Acknowledgements
My sincere thanks go to Jim Hyde and his crew for the exciting time at the National Biomedical EPR Center in Milwaukee, and to all my students, co-workers and collaborators, named in the cited references, for all their valuable contributions. Work in the laboratory was supported by the Deutsche Forschungsgemeinschaft, the National Science Foundation, the Volkswagen-Stiftung, the German–Israel Foundation, and the University of Konstanz.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kroneck, P.M.H. Walking the seven lines: binuclear copper A in cytochrome c oxidase and nitrous oxide reductase. J Biol Inorg Chem 23, 27–39 (2018). https://doi.org/10.1007/s00775-017-1510-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-017-1510-z