
Abstract
Various strategies have been explored in the last 20 years to modify the functional properties of proteins and, among these, protein/polymer conjugation resulted one of the most successful approaches. Thus, the surface modification of polypeptides of potential industrial interest by covalent attachment of different macromolecules is nowadays regarded as an extremely valuable technique to manipulate protein activities. Protein derivatives with a number of either natural or synthetic polymers, like different polysaccharides or polyethylene glycol, have been obtained by both chemical and enzymatic treatments, and in this context, the crosslinking enzyme transglutaminase is attracting an increasing attention as a simple and safe means for protein processing in vitro. In this short review, we summarized the most significant experimental findings demonstrating that a microbial form of the enzyme is an effective tool to obtain several biopolymer-based conjugates potentially useful for both food and pharmaceutical applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein hydrophilic/phobic balance and net charge can be significantly modified by covalent or non-covalent binding of different macromolecules that change protein conformation and functional characteristics, including thermostability, solubility, interfacial properties, degree of hydration and propension to gelation. In particular, protein complexes with different biopolymers, like polysaccharides, perform many noteworthy functions inside the cells but they could play also extremely relevant roles in different biotechnological applications. One of these is to contribute to the structural and textural properties of foods through their aggregation and gelation behavior (Dickinson 2003). By their linking, in fact, proteins and polysaccharides may combine their individual traits and produce food ingredients with a wider range of structural and functional properties. In addition, hydrogels have significant applications in tissue engineering and drug delivery, since single materials are often unable to fully satisfy the medical requirements. Consequently, novel composite hydrogels are always gaining increasing interest (Doumeche et al. 2007; Picard et al. 2009). Furthermore, several proteins obtained in large amounts by recombinant DNA techniques have become effective new drugs (Pavlou and Reichert 2004) but, despite these significant advances, protein drugs often present shortcomings limiting their use, such as low solubility, high susceptibility to proteolysis, marked capacity to generate antibodies, short circulating half-life in vivo and rapid kidney clearance (Harris and Chess 2003; Malik et al. 2007). Therefore, among the various strategies explored to overcome these problems, the conjugation of protein drugs with different natural or synthetic polymers is nowadays considered as a valuable methodology (Harris and Chess 2003; Greenwald et al. 2003; Pasut et al. 2004). Finally, several protein/polysaccharide complexes have been also synthesized to improve the stability properties and to influence kinetics of different purified enzymes of practical interest, as well as to immobilize them, or to prepare biodegradable edible films with desirable mechanical and/or barrier properties to water vapor, CO2 and O2 (Villalonga et al. 2003a, b, 2006a; Valdivia et al. 2006; Di Pierro et al. 2010; Mariniello et al. 2010).
Protein/polysaccharide binding may occur by physical interactions and/or by covalent crosslinking. Complexation pH (pHc) is a significant factor influencing the non-covalent binding between proteins and other macromolecules. During the titration of a polyanion/polycation mixture from high pH, as the charge on the polycation is reduced there is a transition at a specific pH value, called pHc, where a soluble complex is formed (Hattori et al. 2001; Weinbreck and De Kruif 2003). Then, the complex may be further stabilized through other intermolecular forces like hydrophobic ones and/or hydrogen bonds (Hallberg and Dubin 1998; Girard et al. 2002). Attractive interactions between positively (or negatively) charged proteins and anionic (or cationic) biopolymers can lead to gelation, coacervation, or multilayer formation (MacDougall et al. 2001; Turgeon et al. 2003). Consequently, the overall stability and texture of colloidal systems depend not only on the properties of the individual biopolymers, but also on the nature and strength of their interactions. In fact, highly structured protein/polysaccharide complexes may exhibit better functionality—as interfacial, adsorption and hydration properties—than that of the protein or the polysaccharide alone (Ye 2008).
Conversely, polysaccharide covalent binding to proteins may be derived from either chemical or enzyme-catalyzed reactions. In this context, the Maillard reaction is of growing scientific interest mainly to improve solubility and/or emulsifying, gel forming and antioxidant properties of food proteins (Miller and Gerrard 2005). However, this chemical reaction is known to produce some mutagenic compounds (Brands et al. 2000) and undesired browning products (Guerra-Hernandez et al. 2002) and, consequently, may affect sensory attributes of the modified proteins, potentially giving rise also to safety problems.
Another example of chemical protein conjugation is the covalent attachment of polyethylene glycol (PEG), a non-charged and highly hydrophilic polymer used to increase the half-life of polypeptide drugs circulating in blood and to decrease their immunogenicity (Veronese and Mero 2008; Jain and Jain 2008). The most widely employed method for protein PEGylation involves the covalent binding of activated monomethoxy-PEG at level of the ε-amino group of Lys residues using different monomethoxy-PEG derivatives. However, this chemical strategy has strong limitations due to potential multiple sites of conjugation inside the proteins and to the consequent heterogeneity of PEGylated products. Thus, the purification of the proteins modified by this way is often complicated and this drawback decreases the predictability of their activity and the reproducibility of the procedure needed for the required regulatory approval. To reach site-specific PEGylation, different chemical approaches were developed, such as the selective PEGylation either at the level of endoprotein Cys or at their N-terminal amino groups (Zalipsky 1995; Kinstler et al. 2002).
A milder approach to produce protein/polymer conjugates is certainly desirable due to possible toxic side reactions and adverse public opinion on the use of chemicals mainly in food production as well as in medical applications. Accordingly, different enzymatic procedures have been proposed and tested as alternative to the chemical coupling. There are several motivations for employing enzymes for macromolecular processing, the most important of which are enzymes that allow selectivity for the precise coupling of macromolecules and catalyze reactions under biological conditions (Cheng and Gross 2005). Two major enzymatic methods have been used for the conjugation of proteins with other biomacromolecules including polypeptides. The first enzymatic method was focused on tyrosinases, copper-containing phenol oxidases typically responsible for both synthesis of melanin pigments and food browning (Yoruk and Marshall 2003; Mayer 2006). Tyrosinases are known to be able to react with substrates by oxidizing solvent-accessible phenolic residues of endoprotein Tyr, thus activating macromolecular substrates as a means to graft peptides (Aberg et al. 2004; Anghileri et al. 2007), polypeptides (Kang et al. 2004; Freddi et al. 2006) and even synthetic polymers (Shao et al. 1999) to polysaccharides. One of the main limitations of protein tyrosinase-mediated oxidation is that the phenolic residue must be easily accessible. Therefore, the tyrosinase-mediated grafting of globular proteins, having highly compact structures, to other macromolecules generally requires a prior introduction of accessible phenolic substituents (Lewandowski et al. 2008). The second more promising biocatalytic methodology for the conjugation of proteins with a variety of other synthetic and natural polymers exploits the crosslinking activity of the enzyme transglutaminase (EC 2.3.2.13; TGase). TGase, through its ability to produce either isopeptide bonds between endoprotein Gln and Lys residues or γ-glutamyl derivatives with compounds containing primary amino groups has been shown to modify functional properties of both peptides (Esposito et al. 1995) and proteins from different origin (Faergemand et al. 1998; Mariniello et al. 2007a; Ramezani et al. 2008; Sorrentino et al. 2012; Giosafatto et al. 2012; Porta et al. 2013). Multiple molecular forms of TGase are known to play different biological roles inside and outside the cells (Lorand and Graham 2003), representing a large family of enzymes occurring not only in many tissues and body fluids of mammals but also in invertebrates and plants. The most studied molecular form of the enzyme in eukaryotes is the so-called “tissue” TGase or TGase2, a 75-kDa monomeric globular protein expressed in the majority of cells and tissues (Fesus and Piacentini 2002). Although it was shown that all TGases recognize a wide variety of primary amines as acyl acceptor substrate, their specificity for protein bound Gln residues as acyl donor substrates is quite stringent. A number of experiments have been performed, using peptides and proteins, to clarify the pattern of amino acid sequences needed to make reactive a Gln residue. The results of these studies indicated that both primary structure and local conformation around Gln involved in the TGase-catalyzed reaction contribute to determine the enzyme specificity (Coussons et al. 1992) and that in globular proteins, the reactive Gln must be located in protein segments characterized by enhanced chain flexibility (Fontana et al. 2008).
In the late 1980, TGase activity has been detected also in bacteria and some of the genes encoding the enzyme have been successfully cloned (Ando et al. 1989; Washizu et al. 1994). The microbial TGase (mTGase) derived from a variant of Streptomyces mobaraense is constituted by a single polypeptide chain of 331 amino acids with a mol. wt. of 37.9 kDa. Its overall structure consists of a compact domain and Cys-64, the residue essential for the catalytic activity, located at the bottom of a deep cleft (Kashiwagi et al. 2002). Moreover, mTGase exhibits no significant sequence identity with mammalian enzymes, has a broader substrate range and is calcium-independent (Shimba et al. 2002; Yokohama et al. 2004). These features, together with that of a higher reaction rate, are extremely favorable for the exploitation of such molecular form of TGase as a versatile reagent for protein modification in vitro and, consequently, as a biotechnological tool especially for industrial applications. mTGase does not require activated substituents or additional reagents and, therefore, it provides a simple and safe method for coupling small or high molecular weight molecules to protein substrates. In fact, crosslinking reaction catalyzed by mTGase takes place at the enzyme active site offering greater selectivity compared with the one involving tyrosinase, which generates a reactive intermediate that undergoes a non-enzymatic coupling reaction. In addition, mTGase may more easily allow protein conjugation with either fluorescent or radioactive probes without the loss of protein activity, often observed when unspecific labeling or over labeling occurs. As well as it offers the possibility to covalently link carrier proteins to Lys- or Gln-containing substrates of some pharmaceutical interest for specific therapeutic treatments.
Therefore, even though mTGase has been extensively used so far mostly to improve the physical and textural properties of several protein-rich foods (Zhu et al. 1995), this enzyme is now receiving increasingly considerable attention as a tool to couple macromolecules, generating crosslinked networks, and to graft substituents to proteins, tailoring specific activities or conferring hybrid properties (Sato et al. 2001, 2004a, b; Yang et al. 2009). In this review, we summarize the main results obtained so far by using mTGase to site-selectively modify proteins by crosslinking other polymers, often suitably derivatized to serve as enzyme acyl acceptor substrates.
Transglutaminase-catalyzed protein PEGylation
Polymer/drug conjugates are extremely promising candidates for the delivery of a variety of therapeutic agents. Among the synthetic macromolecules so far investigated, PEG is a highly hydrophilic and non-charged polymer demonstrated to be non-toxic when its molecular mass is lower than 1,000 Da and, thus, its use has been allowed by the US FDA (Harris and Chess 2003). The control of drug circulating lifetimes and tissue distribution is one of the main fields to develop new drug delivery systems. In this respect, PEGylation of different pharmaceuticals has been shown to be an effective strategy because PEG conjugates may increase water solubility and stability of the agents and improve their pharmaceutical profile, thereby reducing the possibility of undesirable side effects. Therefore, derivatization by PEG chains is a useful methodology for drug development and is widely exploited to conjugate oligonucleotides, peptides and proteins (Jain and Jain 2008; Veronese and Mero 2008). Various strategies have been investigated to improve the clinical properties of various proteins, by both protein engineering and producing chimeric protein drugs fused to albumin. Among the different approaches, the surface modification of proteins by covalent attachment of PEG is nowadays regarded as an extremely effective technique. In particular, PEGylation is employed to enhance the circulating half-life of therapeutic proteins in blood, to increase the resistance to proteolytic inactivation, and to reduce their antigenicity, immunogenicity and renal clearance (Roberts et al. 2002; Wattendorf and Merkle 2008). Thus, a number of protein/PEG conjugates, such as adenosine deaminase/PEG, asparaginase/PEG, interferon/PEG and PEG-uricase are currently available in the market and found their application in the treatment of severe combined immune deficiency syndrome, acute lymphocytic leukemia, hepatitis C and gout disease, respectively (Duncan and Vicent 2013).
Protein/PEG conjugates are usually prepared by techniques of random derivatization of the endoprotein Lys residues, but this procedure leads to both heterogeneity and decreased bioactivity of the obtained products. Therefore, the development of new methodologies aimed to site-specific protein PEGylation is fundamental to synthesize homogeneous and pharmacologically effective conjugated products for clinical use. Sato et al. (1996) have first conceived an enzymatic method for the site-specific incorporation of PEG-alkylamine derivatives using genetic engineering techniques and TGase2 from guinea pig liver. This methodology appeared more effective than the previously used ones because of both mild reaction conditions and limitation of PEG incorporation to Gln residues, acting as acyl donor substrates of the enzyme. The same authors developed few years later a novel methodology using mTGase as catalyst and recombinant human interleukin 2 (rhIL-2) as target protein together with two model acyl acceptor substrates of the enzyme. The first was a 12-kDa PEG derivative (PEG12), a synthetic polymer endowed with a linear alkylamine chain at one end useful for prolonging protein circulating lifetimes in vivo, whereas the second one was a galactose-terminated triantennary glycoside, (Gal)3, with a linear alkylamine chain at one end (Sato et al. 2000, 2001). (Gal)3 is an artificial ligand for the hepatic asialoglycoprotein receptor, able to recognize exposed and branched galactose residues on serum glycoproteins and utilized as an hepatic targeting device. The derivative peptide mapping, performed by liquid chromatography-electrospray ionization mass spectrometry, showed that only one Gln of the six Gln residues occurring in rhIL-2 was site-specifically modified, thus indicating that only Gln-74 was a reactive acyl donor substrate for mTGase. Moreover, tests of biological activity of PEG12- and (Gal)3-modified rhIL-2 showed that the two conjugates had the same activity of the unmodified protein. In addition, pharmacokinetic studies indicated that PEG12 derivative was eliminated more slowly from the circulation, whereas (Gal)3 derivative accumulated in the liver (Sato et al. 2001). These results, hence, strongly encouraged further investigations for the preparation of other protein/PEG conjugates for clinical use using mTGase. More recently, the mTGase-catalyzed synthesis of the PEG-NH2 derivatives of both apomyoglobin and human growth hormone (hGH) has been reported (Fontana et al. 2008; Mero et al. 2009). Despite there being up to 6 Gln residues in the apomyoglobin molecule, a homogeneous mono-PEGylated protein conjugate was isolated at high yield by chromatography. Fingerprinting techniques combined with mass spectrometric analyses revealed that the enzyme selectively linked PEG-NH2 to a specific Gln residue (Gln-91) of the 153-residue long apomyoglobin chain. A still more interesting result was obtained by the same authors following the derivatization experiments carried out with the hGH in an attempt to synthesize a possible conjugate having a longer functional half-life in vivo to avoid frequent injections of the hormone. As it is well known, hGH is a single polypeptide chain of 191 amino acids with effects not only on the growth but also on glucose metabolism, lactation, and macrophage activation, and its deficiency is generally treated by daily subcutaneous administrations of the hormone. It was demonstrated that, even though hGH contains 13 Gln residues, major conjugated products specifically PEGylated at Gln-40 and/or Gln-141 were obtained in the presence of PEG-NH2 and mTGase. Moreover, enzymatic digestion experiments carried out with both PEGylated apomyoglobin and hGH showed a close correlation between the protein sites recognized by TGase and the ones target of several proteases.
Finally, Maullu et al. (2009) successfully mTGase-PEGylated the granulocyte colony-stimulating factor (GCSF) with the aim to obtain GCSF derivative(s) with longer circulation times in vivo. GCSF, a protein constituted by 174 amino acids and containing 17 Gln residues as potential acyl donor sites for the enzyme, is known to play a physiological role in hematopoiesis by controlling the granulocyte production, differentiation and function, and is also widely used as an effective drug to treat neutropenia. However, since the native protein is rapidly removed from the body by a combination of renal and neutrophil clearance processes, repeated injections or continuous infusion of GCSF are needed to generate sufficiently elevated levels of neutrophils and mobilized progenitor/stem cells in peripheral blood. For these reasons, GCSF-PEGylation might represent an effective strategy to obtain new and more useful therapeutic derivatives. Modified GCSF, homogeneously mono-PEGylated by mTGase at level of Gln-134, seems, thus, of potential clinical interest being expected to have a longer circulating half-life than the wild-type protein. In fact, preliminary pharmacological studies by subcutaneous administration in normal and neutropenic rats showed that enzyme-PEGylated GCSF has the same biological activity of the unmodified protein and better pharmacokinetic parameters (Maullu et al. 2009).
Transglutaminase-catalyzed protein glycosylation
Positive results achieved using TGase to PEGylate proteins suggested the use of the enzyme to selectively couple other polymers with various polypeptide targets, including enzymes, through the functionalization of different biological macromolecules of polysaccharide nature with primary amino groups that act as acyl acceptor substrates for the enzyme. Although Ohtsuka et al. (2000) found that the amino group should be linked to an alkyl chain containing more than four carbon atoms to be an effective mTGase substrate, other experimental routes included the functionalization with N-terminal Gly and Lys- or Gln-containing tags (Tanaka et al. 2005; Tominaga et al. 2007).
Industrial application of enzymes is often limited by their low stability properties under common technological conditions. In this regard, the covalent attachment of ionic and non-ionic polysaccharides such as chitosan, pectin (PEC), dextran (DEX), carboxymethylcellulose (CMC), mannan, alginate (ALG) and cyclodextrin (CD)-grafted polysaccharides has been reported as a useful tool for increasing the stability of various enzymes. In recent decades, a great number of investigations have been devoted to improve functional stability and functionality of the proteolytic enzymes by water-soluble polymer attachment. Since chemical modification has often been reported to provoke significant losses of catalytic activity, the development of new methods for stabilizing proteases, preserving their catalytic properties, received considerable attention. Therefore, we used mTGase as catalyst for the incorporation into the polypeptide structure of pancreatic trypsin of mono-6-amino-6-deoxy derivatives of α-, β- and γ-CD, mono-6-ethylenediamino-, mono-6-propylenediamino-, mono-6-butylenediamino- and mono-6-hexylenediamino-6-deoxy-β-CD, as well as of the aminated derivatives of DEX, CMC and Ficoll (FIC), in an attempt to change the functional properties of the protease (Villalonga et al. 2003a, b, 2006). CDs are capable of forming inclusion complexes with many hydrophobic compounds, and this property has gained favor with wide use in different biomedical and industrial applications. The TGase-synthesized trypsin/CD conjugates, containing about 3 mol of oligosaccharide per mole of protein, exhibited significantly improved specific esterolytic activities and kinetic constants as well as more resistance to both autolytic degradation at alkaline pH and to heat inactivation. DEX (linear and neutral polymer with a mol. wt. of 7.2 × 104), CMC (linear and negatively charged polymer of mol. wt. of 2.5 × 104) and FIC (highly branched and neutral polymer of mol. wt. of 6.9 × 104) were all derivatized with 1,4-diaminobutane prior to be covalently attached to trypsin through mTGase. The TGase-synthesized trypsin/polysaccharide conjugates contained an average of 0.7–1.8 mol of polymers per mol of protein, retained about 61–82 % of the esterolytic activity of the unmodified protease and exhibited an increased stability against both heat treatment and several denaturing conditions. According to these findings, the TGase-mediated covalent derivatization of trypsin aminated either with oligo- or polysaccharides revealed to be a useful strategy for increasing the functional stability of such protease that is widely used in food manufacturing and processing industry.
TGase-catalyzed coupling with aminated-DEX was also utilized to modify liver catalase, an antioxidant enzyme with potential applications in the therapy of several diseases mediated by reactive oxygen species. The beneficial effect of chemical modification of catalase with biocompatible polymers was previously demonstrated by testing a catalase/PEG derivative in rat models of both nephrotic syndrome and lung injury due to asbestosis. The ability of catalase to act as acyl donor substrate of mTGase was preliminarily demonstrated by testing the reactivity of its Gln residue(s) towards monodansylcadaverine, a well-known low mol. wt. amino donor TGase substrate. Then, a catalase/aminated-DEX conjugate, with high purity and well-defined polymer content and mol. wt., was obtained (Valdivia et al. 2006). In contrast to other catalase/polymer conjugates previously synthesized by chemical methods, the adduct prepared by TGase-mediated reaction showed an increased catalase activity in comparison with the unmodified enzyme. In addition, crosslinking of catalase with aminated-DEX significantly enhanced its resistance against both thermal inactivation and tryptic degradation, while on the other hand, improved its pharmacokinetics performance contributing to its higher half-life time and lower clearance in comparison with the unmodified counterpart. However, it should be noted that the observed prolongation of the serum half-life period was lower than that detected with the catalase adduct prepared by chemical conjugation with PEG. This could be a consequence of the low amount of DEX molecules covalently bound to each molecule of catalase, due to the limited number of reactive Gln residue(s) occurring in the catalase polypeptide chain. By contrast, DEX biodegradability and its lower toxicity, the higher catalytic activity exhibited by the prepared conjugate, as well as the benefits derived from the use of an enzyme-catalyzed coupling reaction, all constitute important advantages of the described catalase/aminated-DEX glycoconjugate. Therefore, these findings strongly support the notion that mTGase-catalyzed incorporation of end-group aminated-DEX to reactive endoprotein Gln residues might be a useful tool for preparing protein/polysaccharide prodrugs with specific pharmacological applications.
Also aminated-PEC and aminated-ALG were demonstrated to be effective acyl acceptor substrates of mTGase and, hence, potentially used to synthesize further protein/polysaccharide conjugates. In fact, we recently linked 1,4-diaminobutane (putrescine, PT) both to ALG and low-methoxyl PEC to synthesize new aminated polysaccharides and demonstrated that both PT-PEC and PT-ALG are effective amino donors for the enzyme in vitro using dimethylated casein and soy flour proteins as acyl donor TGase substrates (Di Pierro et al. 2010). Thus, we covalently coupled the two PT-polysaccharides to soy flour proteins by means of the enzyme and prepared edible films. Characterization of the crosslinked films showed a significantly decreased water vapor permeability, as well as improved mechanical properties such as high extensibility, with respect to the control films where protein and polysaccharide molecules were not covalently bound to each other. These results open, in our opinion, a new chapter in the biodegradable/edible polymer field finalized to obtain hydrocolloid films with marked extensibility and high-moisture barrier capacity for potential applications not only in food industrial sector but also in pharmaceutical and agricultural ones.
Finally, hydroxyethyl starch (HES), a semisynthetic biodegradable polymer widely used as a blood plasma volume expander, has also attracted the attention as a promising PEG substitute. HES exhibits high water solubility and low hypersensitivity, as well as allows the possibility to tailor its molar mass and biodegradation rate. Besheer et al. (2009) reported the modification of HES with both hexamethylene diamine and N-carbobenzyloxyglutaminyl-glycine (Z-Gln-Gly) to render HES a reactive acyl acceptor and donor substrate, respectively, for a highly purified recombinant mTGase with 6 His residues at the C-terminus (Marx et al. 2008). This enzymatic procedure was demonstrated to be a further feasible and promising strategy for the development of methods alternative to the well-established protein PEGylation, a simple and mild approach for the possible conjugation of polypeptides to fully biodegradable and water-soluble polymers with the aim to alter pharmacokinetics and biological fate of protein drugs.
Figure 1 schematically shows all the demonstrated crosslinked conjugates synthesized in vitro in the presence of TGase.

Crosslinked conjugates synthesized in vitro in the presence of TGase
Non-covalent transglutaminase-crosslinked protein/polysaccharide complexes
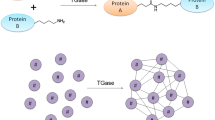
Non-covalent protein/polysaccharide complexes, potentially useful for both food and pharmaceutical applications, were also obtained by mTGase-catalyzed protein/protein crosslinking. Therefore, the manipulation of the attractive interactions between charged proteins and polysaccharides interactions (Dickinson 2008) can represent an important tool to modify the microstructure and the shelf-life of the composite systems mostly in the edible films, since the formation of a continuous network strictly depends on the biopolymer behavior in the film-forming solution. A promising exploitation of mTGase in this field is related to the production of the so-called “bioplastics”. In fact, although most of the biodegradable and/or edible plastics produced so far include polymers of mineral origins, such as polyesters and polyvinyl alcohols, those of natural origin contain polysaccharides, proteins, lipids and polyesters synthesized by several microorganisms (Bourtoom 2008). Films prepared with polysaccharides are quite resistant but exhibit poor water vapor barrier features, owing to their hydrophilic nature, whereas protein-based films show superior oxygen barrier characteristics. Therefore, a great interest was devoted to investigate the possibility to introduce crosslinks into the film network “enzymatically” and, thus, mTGase is stimulating an increased deal of interest as potential tool in reticulating proteins of hydrocolloid multi-component edible films to obtain bioplastics with desired mechanical and gas barrier properties (Porta et al. 2011a). In particular, we produced films of either PEC or chitosan in the presence of different plant and milk proteins that act as mTGase substrates, and we demonstrated that the enzyme-catalyzed formation of protein crosslinks into the film network generally determines a decreased solubility of the bioplastics and increases its capacity to stretch (Porta et al. 2011b). Specifically concerning the PEC-based materials, the effect of mTGase was shown both to significantly increase film barrier properties to both O2 and CO2 and to confer a moderate film permeability to water vapor. These modifications of bioplastics characteristics suggest their possible use in a variety of situations, one of which could be the edible wrapping of high-moisture food to prevent their quality changes. In fact, in a recent study, we prepared whey protein/PEC films, crosslinked or not by mTGase, to obtain possible hydrocolloid food coatings with appropriate features (Di Pierro et al. 2013). Our findings indicated that the enzyme had a considerable influence on whey protein/PEC complex formation and, as a consequence, on the mechanical and barrier properties of the obtained edible films. In particular, we demonstrated the formation at pH 5.1 (pHc) of mTGase-catalyzed crosslinks among soluble ionic whey protein/PEC complexes, which could be responsible for the observed increase of both tensile strength (2-fold) and elongation to break (10-fold) of films obtained in the presence of the enzyme. Furthermore, the protein crosslinking catalyzed by mTGase at pHc was also shown to significantly reduce film permeability, this effect being probably due to the marked decrease of the porosity of whey protein/PEC films observed by both atomic force and scanning electron microscopy. A schematic representation of non-covalent protein/polysaccharide complexes obtained by mTGase-catalyzed protein/protein crosslinking at pHc is shown in Fig. 2.

Non-covalent protein/polysaccharide complexes obtained following TGase-catalyzed protein/protein crosslinking at pHc
As far as the possible application outcomes, we demonstrated that food coating with an edible whey protein/PEC film prepared in the presence of the enzyme proved to be an effective way to significantly decrease oil content in widely consumed deep-fat fried foods (doughnuts and French fries) and to prevent water absorption from baked foods, like “Taralli”, over time (Rossi Marquez et al. 2013). In this respect, it is well known that minimizing water desorption/absorption in both fried and baked foods is of wide interest because moisture plays a crucial role in determining their quality and shelf-life. The reported methodology thus suggested producing healthier fried foods and avoiding the baked ones that quickly become soggy and soft by losing their hardness and crispness.
A possible application in the agricultural field of PEC-based bioplastics is represented by that produced with fennel waste and containing in its matrix mTGase-crosslinked phaseolin, a seed storage protein of the common bean. The latter material was suggested as a promising candidate for the production of an environmentally friendly sheet mulching (Mariniello et al. 2007b). In addition, a significant improvement in the mechanical resistance of TGase-crosslinked bioplastics was observed using chitosan in the presence of ovalbumin and whey proteins (Di Pierro et al. 2006, 2007). In fact, protein crosslinking inside the chitosan network seems to reduce the intermolecular chain mobility of the polysaccharide matrix, thus increasing the tensile strength and reducing the extensibility of the produced bioplastics. The reported marked decrease in chitosan-based film permeability to O2 and CO2, as well as the lower water vapor permeability, are probably associated with the resulting more compact structure of such biomaterial and with the changes in its hydrophilic properties. However, sustained multidisciplinary researches are necessary both to exploit the hitherto produced crosslinked bioplastics for specific industrial applications and to develop new eco-friendly materials to widen their potential use. Thus, the possibility to obtain “tailored” molecular networks conferring desired features to specific bioplastics with the aid of TGase remains an attractive perspective in this field of investigations.
Finally, a casein/konjac glucomannan (KGM) hydrogel was recently obtained in the presence of mTGase and reported as a non-toxic and biodegradable protein/polysaccharide conjugate with potential biomedical applications (Yin et al. 2012). Biocompatible hydrogels, being able to absorb water several times their own weight, are another attractive new materials to be used as drug vectors. Generally, single-material hydrogels do not fully satisfy the requirements of efficient drug delivery systems and, consequently, novel composite hydrogels are gaining increased interest. Therefore, KGM, a high mol. wt., water-soluble and water-absorbent natural polysaccharide, was proposed as a stabilizer of mTGase-crosslinked casein hydrogels. Since KGM is selectively degraded by β-mannanase in the colon, its use was suggested in colon-specific drug delivery systems. In this respect, successful preliminary experiments were carried out by oral administration of hydrogel-incorporated docetaxel, a hydrophobic antineoplastic agent known to have undesirable side effects when administered alone by injection.
References
Aberg CM, Chen TH, Olumide A, Raghavan SR, Payne GF (2004) Enzymatic grafting of peptides from casein hydrolysate to chitosan. Potential for value-added byproducts from food-processing wastes. J Agric Food Chem 52:788–793
Ando H, Adachi M, Umeda K, Matsuura A, Nonaka M, Uchio R, Tanaka H, Motoki M (1989) Purification and characterization of a novel transglutaminase derived from microorganisms. Agric Biol Chem 53:2613–2617
Anghileri A, Lantto R, Kruus K, Arosio C, Freddi G (2007) Tyrosinase-catalyzed grafting of sericin peptides onto chitosan and production of protein–polysaccharide bioconjugates. J Biotechnol 127:508–519
Besheer A, Hertel TC, Kressler J, Mader K, Pietzsch M (2009) Enzymatically catalyzed HES conjugation using microbial transglutaminase: proof of feasibility. J Pharm Sci 98:4420–4428
Bourtoom T (2008) Edible films and coatings: characteristics and properties. Int Food Res J 15:1–12
Brands CMJ, Alink GM, Van Boekel MAJS, Jongen WMF (2000) Mutagenicity of heated sugar-casein systems: effect of the Maillard reaction. J Agric Food Chem 48:2271–2275
Cheng HN, Gross RA (2005) Polymer Biocatalysis and Biomaterials. ACS, Washington, DC
Coussons PJ, Price NC, Kelly SM, Smith B, Sawyer L (1992) Factors that govern the specificity of transglutaminase-catalysed modification of proteins and peptides. Biochem J 282:929–930
Di Pierro P, Chico B, Villalonga R, Mariniello L, Damiao AE, Masi P, Porta R (2006) Chitosan-whey protein edible films produced in the absence or presence of transglutaminase: analysis of their mechanical and barrier properties. Biomacromolecules 7:744–749
Di Pierro P, Chico B, Villalonga R, Mariniello L, Masi P, Porta R (2007) Transglutaminase-catalyzed preparation of chitosan-ovalbumin films. Enzyme Microb Tech 40:437–441
Di Pierro P, Mariniello L, Sorrentino A, Villalonga R, Chico B, Porta R (2010) Putrescine–polysaccharide conjugates as transglutaminase substrates and their possible use in producing crosslinked films. Amino Acids 38:669–675
Di Pierro P, Rossi Marquez G, Mariniello L, Sorrentino A, Villalonga R, Porta R (2013) Effect of transglutaminase on the mechanical and barrier properties of whey protein/pectin films prepared at complexation pH. J Agric Food Chem, in press
Dickinson E (2003) Hydrocolloids at interfaces and the influence on the properties of dispersed systems. Food Hydrocoll 17:25–39
Dickinson E (2008) Interfacial structure and stability of food emulsions as affected by protein–polysaccharide interactions. Soft Matter 4:932–942
Doumeche B, Picard J, Larreta-Garde W (2007) Biomacromolecules 8:3613–3618
Duncan R, Vicent MJ (2013) Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev 65:60–70
Esposito C, Mancuso F, Calignano A, Di Pierro P, Pucci P, Porta R (1995) Neurokinin receptors could be differentiated by their capacity to respond to the transglutaminase-synthesized γ-(glutamyl5)spermine derivative of substance P. J Neurochem 65:420–426
Faergemand M, Otte J, Qvist KB (1998) Cross-linking of whey proteins by enzymatic oxidation. J Agric Food Chem 46:1326–1333
Fesus L, Piacentini M (2002) Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci 27:534–539
Fontana A, Spolaore B, Mero A, Veronese FM (2008) Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv Drug Delivery Rev 60:13–28
Freddi G, Anghileri A, Sampaio S, Buchert J, Monti P, Taddei P (2006) Tyrosinase-catalyzed modification of Bombyx mori silk fibroin: grafting of chitosan under heterogeneous reaction conditions. J Biotechnol 125:281–294
Giosafatto CVL, Rigby NM, Wellner N, Ridout M, Husband F, Mackie AR (2012) Microbial transglutaminase-mediated modification of ovalbumin. Food Hydrocoll 26:261–267
Girard M, Turgeon SL, Gauthier SF (2002) Inter biopolymer complexing between beta-lactoglobulin and low- and high-methylated pectin measured by potentiometric titration and ultrafiltration. Food Hydrocoll 16:585–591
Greenwald R, Choe Y, McGuire J, Conover C (2003) Effective drug delivery by PEGylated drug conjugates. Adv Drug Delivery Rev 55:217–250
Guerra-Hernandez E, Gomez CL, Garcia-Villanova B, Sanchez NC, Gomez JMR (2002) Effect of storage on non-enzymatic browning of liquid infant milk formulae. J Sci Food Agric 82:587–592
Hallberg RK, Dubin PL (1998) Effect of pH on the binding of beta-lactoglobulin to sodium polystyrene sulfonate. J Phys Chem 102:8629–8633
Harris JM, Chess R (2003) Effect of PEGylation on pharmaceuticals. Nature Rev Drug Discov 2:214–221
Hattori T, Kimura K, Seyrek E, Dubin PL (2001) Binding of bovine serum albumin to heparin determined by turbidimetric titration and frontal analysis continuous capillary electrophoresis. Anal Biochem 295:158–167
Jain A, Jain SK (2008) PEGylation: an approach for drug delivery. A review. Crit Rev Ther Drug Carrier Syst 25:403–447
Kang GD, Lee KH, Ki CS, Nahm JH, Park JH (2004) Silk fibroin/chitosan conjugate crosslinked by tyrosinase. Macromol Res 12(534):539
Kashiwagi T, Yokoyama K, Ishikawa K, Ono K, Ejima D, Matui H, Suzuki E (2002) Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense. J Biol Chem 277:44252–44260
Kinstler O, Molineux G, Treuheit M, Ladd D, Gegg C (2002) Mono-N-terminal poly(ethylene glycol)–protein conjugates. Adv Drug Deliv Rev 54:477–485
Lewandowski AT, Yi HM, Luo XL, Payne GF, Ghodssi R, Rubloff GW, Bentley WE (2008) Protein assembly onto patterned microfabricated devices through enzymatic activation of fusion pro-tag. Biotechnol Bioeng 99:499–507
Lorand L, Graham RM (2003) Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4:140–156
MacDougall AJ, Brett GM, Morris VJ, Rigby NM, Ridout MJ, Ring SG (2001) The effect of peptide behaviour–pectin interactions on the gelation of a plant cell wall pectin. Carbohydr Res 335:115–126
Malik DK, Baboota S, Ahuja A, Hasan S, Ali J (2007) Recent advances in protein and peptide drug delivery systems. Curr Drug Deliv 4:141–151
Mariniello L, Giosafatto CVL, Di Pierro P, Sorrentino A, Porta R (2007a) Synthesis and resistance to proteolysis of transglutaminase-crosslinked phaseolin, the major storage protein from Phaseolus vulgaris. J. Agr Food Chem 55:4717–4721
Mariniello L, Giosafatto CVL, Moschetti G, Aponte M, Masi P, Sorrentino A, Porta R (2007b) Fennel waste-based films suitable for protecting cultivations. Biomacromolecules 8:3008–3014
Mariniello L, Giosafatto CVL, Di Pierro P, Sorrentino A, Porta R (2010) Swelling, mechanical and barrier properties of albedo-based films prepared in the presence of phaseolin crosslinked or not by transglutaminase. Biomacromolecules 11:2394–2398
Marx CK, Hertel TC, Pietzsch M (2008) Purification and activation of a recombinant histidine-tagged protransglutaminase after soluble expression in E. coli and partial characterization of the active enzyme. Enzyme Microb Technol 42:568–575
Maullu C, Raimondo D, Caboi F, Giorgetti A, Sergi M, Valentini M, Tonon G, Tramontano A (2009) Site-directed enzymatic PEGylation of the human granulocyte colony-stimulating factor. FEBS J 276:6741–6750
Mayer AM (2006) Polyphenol oxidases in plants and fungi: going places? A review. Phytochemistry 67:2318–2331
Mero A, Spolaore B, Veronese FM, Fontana A (2009) Transglutaminase-mediated PEGylation of proteins: direct identification of the sites of protein modification by mass spectrometry using a novel monodisperse PEG. Bioconjugate Chem 20:384–389
Miller AG, Gerrard JA (2005) The Maillard reaction and food protein crosslinking. Prog Food Biopolymer Res 1:69–86
Ohtsuka T, Sawa A, Kawabata R, Nio N, Motoki M (2000) Substrate specificities of microbial transglutaminase for primary amines. J Agric Food Chem 48:6230–6233
Pasut G, Guiotto A, Veronese F (2004) Protein, peptide and non-peptide drug PEGylation for therapeutic applications. Expert Opin Ther Pat 14:1–36
Pavlou A, Reichert J (2004) Recombinant protein therapeutics: success rates, market trends and values to 2010. Nat Biotechnol 22:1513–1519
Picard J, Giraudier S, Larreta-Garde V (2009) Soft Matter 5:4198
Porta R, Di Pierro P, Sorrentino A, Mariniello L (2011a) Promising perspectives for transglutaminase in “bioplastics” production. J Biotechnol Biomat 1:3. http://dx.doi.org/10.4172/2155-952X.1000102e
Porta R, Mariniello L, Di Pierro P, Sorrentino A, Giosafatto CVL (2011b) Transglutaminase crosslinked pectin and chitosan-based edible films: a review. Crit Rev Food Sci Nutr 51:1–16
Porta R, Giosafatto CVL, Di Pierro P, Sorrentino A, Mariniello L (2013) Transglutaminase-mediated modification of ovomucoid. Effect on its trypsin inhibitory activity and antigenic properties. Amino Acids 44:285–292
Ramezani R, Esmailpour M, Aminlari M (2008) Effect of conjugation with glucosamine on the functional properties of lysozyme and casein. J Sci Food Agric 88:2730–2737
Roberts MJ, Bentley MD, Harris JM (2002) Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev 54:459–476
Rossi Marquez G, Di Pierro P, Esposito M, Mariniello L, Porta R (2013) Application of transglutaminase-crosslinked whey protein/pectin films as water barrier coatings in fried and baked foods. Food Bioproc Technol. doi:10.1007/s11947-012-1045-9
Sato H, Ikeda M, Suzuki K, Hirayama K (1996) Site-specific modification of interleukin-2 by the combined use of genetic engineering techniques and transglutaminase. Biochemistry 35:13072–13080
Sato H, Yamamoto K, Hayashi E, Takahara Y (2000) Transglutaminase-mediated dual and site-specific incorporation of poly(ethylene glycol) derivatives into a chimeric interleukin-2. Bioconjugate Chem 11:502–509
Sato H, Hayashi E, Yamada N, Yatagai M, Takahara Y (2001) Further studies on the site-specific protein modification by microbial transglutaminase. Bioconjugate Chem 12:701–710
Sato M, Furuike T, Sadamoto R, Fujitani N, Nakahara T, Niikura K, Monde K, Kondo H, Nishimura S (2004a) Glycoinsulins: dendritic sialyloligosaccharide-displaying insulins showing a prolonged blood-sugar-lowering activity. J Am Chem Soc 126:14013–14022
Sato M, Sadamoto R, Niikura K, Monde K, Kondo H, Nishimura S (2004b) Site-specific introduction of sialic acid into insulin. Angew Chem Int Ed Engl 43:1516–1520
Shao LH, Kumar G, Lenhart JL, Smith PJ, Payne GF (1999) Enzymatic modification of the synthetic polymer polyhydroxystyrene. Enzyme Microb Technol 25:660–668
Shimba N, Yokoyama Y, Suzuki E (2002) J Agric Food Chem 50:1330–1334
Sorrentino A, Giosafatto CVL, Sirangelo I, De Simone C, Di Pierro P, Porta R, Mariniello L (2012) Higher susceptibility to amyloid fibril formation of the recombinant ovine prion protein modified by transglutaminase. Biochim Biophys Acta 1822:1509–1515
Tanaka T, Kamiya N, Nagamune T (2005) N-terminal glycine-specific protein conjugation catalyzed by microbial transglutaminase. FEBS Lett 579:2092–2096
Tominaga J, Kemori Y, Tanaka Y, Maruyama T, Kamiya N, Goto M (2007) An enzymatic method for site-specific labeling of recombinant proteins with oligonucleotides. Chem Comm pp 401–403
Turgeon SL, Beaulieu M, Schmitt C, Sancez C (2003) Protein polysaccharide interactions: phase ordering kinetics thermodynamic and structural aspects. Curr Opin Colloid Interface Sci 8:401–414
Valdivia A, Villalonga R, Di Pierro P, Perez YP, Mariniello L, Gomez L, Porta R (2006) Transglutaminase-catalyzed site-specific glycosidation of catalase with aminated dextran. J Biotechnol 122:326–333
Veronese FM, Mero A (2008) The impact of PEGylation on biological therapies. Biodrugs 22:315–329
Villalonga R, Fernandez M, Fragoso A, Cao R, Mariniello L, Porta R (2003a) Thermal stabilization of trypsin by enzymic modification with β-cyclodextrin derivatives. Biotechnol Appl Biochem 38:53–59
Villalonga R, Fernandez M, Fragoso A, Cao R, Di Pierro P, Mariniello L, Porta R (2003b) Transglutaminase-catalyzed synthesis of trypsin-cyclodextrin conjugates. Kinetics and stability properties. Biotechnol Bioeng 81:732–737
Villalonga ML, Villalonga R, Mariniello L, Gomez L, Di Pierro P, Porta R (2006) Transglutaminase-catalysed glycosidation of trypsin with aminated polysaccharides. World J Microbiol Biotechnol 22:595–602
Washizu K, Ando K, Koiked S, Hiros S, Matsuura A, Akagi H, Motoki M, Takeuchi K (1994) Molecular cloning of the gene for microbial transglutaminase from Streptoverticillium and its expression in Streptomyces lividans. Biosci Biotechnol Biochem 58:82–87
Wattendorf U, Merkle HP (2008) PEGylation as a tool for the biomedical engineering of surface modified microparticles. J Pharm Sci 97:4655–4669
Weinbreck F, De Kruif CG (2003) Complex coacervation of globular proteins and gum Arabic. In: Dickinson E, van Vliet T (eds) Food colloids biopolymers and materials. Royal Society of Chemistry, Cambridge, pp 337–344
Yang X, Liu Y, Payne GF (2009) Crosslinking lessons from biology: enlisting enzymes for macromolecular assembly. J Adhes 85:576–589
Ye A (2008) Complexation between milk proteins and polysaccharides via electrostatic interaction: principles and applications: a review. Int J Food Sci Technol 43:406–415
Yin W, Su R, Qi W, He Z (2012) A casein-polysaccharide hybrid hydrogel cross-linked by transglutaminase for drug delivery. J Mater Sci 47:2045–2055
Yokohama K, Nio N, Kikuchi Y (2004) Properties and applications of microbial transglutaminases. Appl Microbiol Biotechnol 64:447–454
Yoruk R, Marshall MR (2003) Physicochemical properties and function of plant polyphenol oxidase: a review. J Food Biochem 27:361–422
Zalipsky S (1995) Functionalized poly(ethylene glycol) for preparation of biologically relevant conjugates. Bioconjugate Chem 6:150–165
Zhu Y, Rinzema A, Tramper J, Bol J (1995) Microbial transglutaminases: a review of its production and application in food processing. Appl Microbiol Biotechnol 44:277–282
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mariniello, L., Porta, R., Sorrentino, A. et al. Transglutaminase-mediated macromolecular assembly: production of conjugates for food and pharmaceutical applications. Amino Acids 46, 767–776 (2014). https://doi.org/10.1007/s00726-013-1561-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1561-6