
Abstract
In recent years, enterovirus A71 (EV-A71) infection has become a major global public health problem, especially for infants and young children. The results of epidemiological research show that EV-A71 infection can cause acute hand, foot, and mouth disease (HFMD) and complications of the nervous system in severe cases, including aseptic pediatric meningoencephalitis, acute flaccid paralysis, and even death. Many studies have demonstrated that EV-A71 infection may trigger a variety of intercellular and intracellular signaling pathways, which are interconnected to form a network that leads to the innate immune response, immune escape, inflammation, and apoptosis in the host. This article aims to provide an overview of the possible mechanisms underlying infection, signaling pathway activation, the immune response, immune evasion, apoptosis, and the inflammatory response caused by EV-A71 infection and an overview of potential therapeutic strategies against EV-A71 infection to better understand the pathogenesis of EV-A71 and to aid in the development of antiviral drugs and vaccines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hand, foot, and mouth disease (HFMD) usually occurs in children under age 5 between April and June and October or December, with the main symptoms being fever and herpes on the hands, feet, mouth, and sometimes other parts of the body, a general feeling of illness, and low-grade fever in the early stage [1]. More importantly, in severe cases, complications can develop that involve the nervous system, such as encephalitis, meningoencephalitis, and acute flaccid paralysis [2]. HFMD is highly correlated with enterovirus (EV) infection. Enteroviruses are single positive-stranded ribonucleic acid (RNA) viruses belonging to the family Picornaviridae, and the main pathogenic serotypes include coxsackievirus (CV) A groups 4–7, 9, 10, and 16 and CV-B groups 1–3, and 5, some serotypes of echovirus, and EV-A71 [3]. EV-A71 and coxsackievirus A16 (CV-A16) have been regarded as the major causes of HFMD, but coxsackievirus A6 (CV-A6) has steadily become one of the main viruses causing HFMD outbreaks in Europe, America, and Asia, including China, Japan, Taiwan, and Thailand [4]. EV-A71 is a highly contagious virus that is primarily transmitted via the fecal-oral route, respiratory droplets, and close contact, and most severe cases and deaths are caused by EV-A71. Although enterovirus D68 (EV-D68) can also cause HFMD, it is spread more similarly to rhinoviruses through the respiratory tract [5].
EV-A71 was first isolated from a child with encephalitis in California in 1969 [6], and EV-A71 infection has been linked to many outbreaks of HFMD worldwide, particularly large outbreaks in the Asia-Pacific region, resulting in a significant number of deaths since 1997 [7,8,9,10,11,12,13,14,15,16]. Since 2000, EV-A71 has been involved in 13 outbreaks in several regions of the world [17], including Japan [18, 19], Australia [20], Malaysia [8], and Taiwan [11]. In 2008, an outbreak of EV-A71 in China prompted the authorities to put the country on alert, with the number of registered cases exceeding 60,000, including 38 deaths [21]. Different subgenotypes of EV-A71 have been found in different regions. For example, subgenotype C4 is predominantly found in China; C2, C4, B4, and B5 are in Taiwan; and C2, C4, and B5 are in Japan [22,23,24,25]. A report from WHO indicated [26] a surge in reported cases in China and Singapore in 2018. In China, 377,629 cases and 4 deaths were reported in July 2018, representing a 27% increase from the same period in 2017, while Singapore reported a cumulative total of 32,956 cases in 2018, with 17,169 requiring hospitalization, but no fatalities. As a result, some Asian countries have already established national surveillance systems for HFMD/EV-A71 [27].
The pathogenesis of EV-A71 infection is intricate and begins with viral attachment to host cells, for which EV-A71 uses scavenger receptor B2 (SCARB2) and other attachment receptors as entry receptors to initiate infection in mammalian cells. This triggers an innate immune response in the host cell, which is initiated when conserved pathogen-associated molecular patterns (PAMPs) are recognized by pattern recognition receptors (PRRs) [28]. However, host immune stress may lead to EV-A71-induced immune avoidance – a process in which viral proteins play a major role – resulting in viral transmission within the host.
EV-A71-induced apoptosis pathways, which include mitochondria-mediated intrinsic apoptosis pathways [29, 30] and non-caspase-dependent pathways caused by Ca2+ influx [31, 32], can lead to irreversible damage to organ tissues on a large scale. In addition, the inflammatory response triggered by EV-A71 infection can lead to inflammation of the nervous system, which can be mediated not only by TLR and RLR receptors [33] but also by EGF receptors [34], which can initiate the MAPK signaling pathway to trigger the release of pro-inflammatory factors [35,36,37,38,39,40,41,42]. Additionally, the accumulation of reactive oxygen species (ROS) in cells and mitochondria caused by EV-A71 also aggravate the inflammatory responses [43].
At present, there are no known effective treatments for EV-A71 infection. Ribavirin and interferon are clinical antiviral drugs that are frequently used, with their mechanism of action mainly centering around inhibiting viral RNA replication and protein translation and aiding immune regulation [44,45,46,47,48,49,50]. More importantly, there are drugs that have not yet been used in the clinic, such as vemurafenib, flavonoids, and nucleic acid aptamer, whose main mechanism is to suppress the inflammatory response pathways [51, 52] or target the viral protein VP1 [53]. Direct-acting EV-A71 antivirals have a site-specific effect. These drugs are promising, and it is expected that they will be used in clinical applications. However, even if effective drugs become available, the most effective way to prevent and control EV-A71 infection is vaccination. Currently, the use of an inactivated EV-A71 vaccine is a preventive measure against outbreaks of HFMD [54,55,56,57,58], while a live attenuated vaccine [59, 60], EV-A71 virus-like particles (VLPs) [61, 62], and a VP1-based synthetic peptide vaccine [63] are still undergoing preclinical evaluation. Each type of vaccine and medication has its advantages and disadvantages, depending on the target group, necessitating individualized treatment for different patients and further research to minimize toxic side effects and treatment limitations.
Below, we review the pathogenesis of EV-A71, focusing on the mechanism by which viruses invade host cells, release RNA to trigger innate immune responses, achieve immune escape through viral proteins, and trigger cellular inflammation and apoptosis pathways. The therapeutic mechanism of drugs for the treatment of EV-A71 infection and the existing vaccines, including their limitations, are also discussed. It is hoped that this review will provide new ideas for therapeutic targets and new insights for exploring more-targeted treatment methods that are suitable for children and have fewer side effects and complications than existing treatment.
Infection
Viral entry
Host receptors
Humans are the natural hosts of EV-A71 [64]. EV-A71 exhibits broad tissue tropism due to its ability to use different cellular receptors for host cell entry. It has been reported that human scavenger receptor class B, member 2 (hSCARB2) is the only receptor known to initiate conformational changes that lead to viral RNA uncoating in the cytoplasm [65, 66], while other molecules facilitate viral attachment to the cell surface without initiating uncoating. For example, heparan sulfate (HS) proteoglycans [67], P-selectin glycoprotein ligand 1 (PSGL-1) [68], sialylated glycan [69, 70], annexin II (Anx2) [71], vimentin [72], nucleolin [73], fibronectin [74], and prohibitin [12] function as “attachment receptors” by retaining the virus on the cell surface.
EV-A71 enters cells by utilizing SCARB2 and attachment receptors, both of which are expressed on the cell surface. The virus binds to these receptors directly, and the host cell then ingests the virus via endocytosis. Once inside the cell, the internalized virus may come into contact with SCARB2 in endosome, and acidification of the endosome causes a rearrangement of the viral capsid structure, leading to the release of viral RNA from the particles, allowing it to be replicated and translated [75]. Blocking the binding of EV-A71 to attachment receptors is a key therapeutic strategy for treating EV-A71 infection. In recent years, antibody-based drugs, such as the monoclonal antibodies A9 and D9, carbohydrates, heparin, and heparin analogs have shown potential for preventing virus attachment and internalization [76].
Factors that facilitate viral invasion
Prohibitins and peripherin
Prohibitins (PHBs) are expressed in nuclei, mitochondria, and the cell membrane, and they play a role in transcription, nuclear signaling, and maintaining the structural integrity of mitochondria [77]. PHBs have been reported to be involved in the mechanism of viral infection, and PHBs expressed on the cell surface are specifically involved in the entry of EV-A71 into neuronal cells, while membrane-bound mitochondrial PHBs are associated with viral replication complexes and promote viral replication. The role of PHBs in the entry of EV-A71 and replication is limited to neuronal-derived cells, supporting the role of PHBs in the neuropathogenesis of EV-A71.
Peripherin (PRPH) also plays a similar role. In motor-neuron-like and neuroblastoma cell lines, surface-expressed PRPH facilitates viral entry, while intracellular PRPH influences viral genome replication through interactions with structural and non-structural viral components [78].
Heat shock protein 90β
When EV-A71 particles attach to the hSCARB2 or PSGL-1, heat shock protein 90β (HSP90β) on the cell surface may act as a cofactor, interacting with the virus to protect its protein from proteasome degradation and contributing to viral assembly and replication. Tsou et al. [79] inhibited EV-A71 entry and subsequent viral replication by knocking down heat shock protein 90 (HSP90) in host cells or targeting HSP90, using the inhibitor geldanamycin (GA) and its analogue 17-allyamino-17-demethoxygeldanamycin (17-AAG). Those experiments also showed that, after EV-A71 particles attach to surface receptors (such as hSCARB2 or PSGL-1), heat shock protein 90 (HSP90) expressed on the cell surface can bind to EV-A71 and facilitate its entry.
Intracellular replication
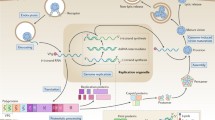
SCARB2 is distributed on both lysosomal and cell membranes. As mentioned above, EV-A71 is capable of binding directly to SCARB2 or other attachment receptors. During infection, EV-A71 can attach to the surface of the host cell and bind to specific receptors with the help of attachment receptors. A change in the structure of the virus particle then results in the loss of VP4, eventually leading to structural changes in the capsid protein, causing pores to form in the host cell membrane and allowing EV-A71 to enter the cell via endocytosis. The virus must then undergo uncoating through a further conformational change to release the viral genome, and this is thought to happen in a SCARB2-dependent and low-pH-dependent manner. However, attachment receptors do not trigger conformational changes of the virus. Internalized viruses that are not bound to SCARB2 may encounter SCARB2 in endosomes and release RNA into the cytoplasm through uncoating after acidification. The viral genomic RNA acts directly as a messenger RNA and is translated into a large polypeptide that is rapidly cleaved by viral proteases into 11 mature structural and non-structural proteins.
The replication of EV-A71 depends on the hijacking of the host translation machinery by subverting cellular cap-dependent translation. The 2A protein encoded by EV-A71 cleaves the host translation initiation factor eIF4G, depriving it of its binding structural domain, which results in the disruption of the cell’s original cap-dependent translation machinery. The 5’ untranslated region (5'UTR) of EV-A71 then binds to a uridylated genome-linked viral protein (VPg) to form an RNA secondary structure, which serves as an internal ribosomal entry site (IRES) that mediates initiation of translation. Cleaved eIF4G recognizes the IRES and recruits ribosomes to translate viral proteins by interacting with the initiation factor eIF3 [80]. The non-structural protein 3D is an RNA-dependent RNA polymerase (RdRp) that mediates replication of the viral genome within a vesicle membrane structure (viral replication complex) [81]. A trilobal structure in the 5’UTR of EV-A71 is a cis-acting element that is necessary for negative chain synthesis and, by binding PABP, VPg, Poly(A), and other components [82], forming a cyclic ribonucleoprotein complex that initiates RNA synthesis and binds to RdRp for viral replication. The viral RNA is copied into negative-stranded RNA, which is then used as a template to synthesize large amounts of positive-stranded RNA [83].
When a large number of viral genome molecules and capsid proteins assemble into mature viral particles, the host cell ruptures and mature viral particles are released. In addition, some studies have shown that EV-A71 particles can be transmitted within secreted exosomes. Huang et al. [84] showed that EV-A71 infection caused differentiated C2BBe1 cells and intestinal organoids to secrete exosomes that contain viral components and have the ability to establish active infection. As the virus is mainly transmitted to peripheral tissues through the oral-gut axis and the exosomes contain contagious viral particles that can infect cells in the intestine, suppressing exosome transmission is essential for the containment of infection in the early stage.
Invasion of the central nervous system
It is hypothesized that primary viral replication occurs in lymphoid tissue of the oropharyngeal cavity (tonsils) and small intestine (Payer’s patch), with further replication taking place in regional lymph nodes (deep cervical and mesenteric nodes), leading to viremia [85]. Sun et al. found that EV-A71 can initially infect the lungs of severely immunocompromised mice, more specifically, type II pneumocytes, and then systematically replicate and infect other organs [86]. The initial colonization site of EV-A71 is still a matter of debate, but if the initial infection is not contained, the virus can spread through the lymphatic circulation to deep lymph nodes and then spread through the blood to other organs or tissues. The receptors PSGL-1 and SCARB2 have been shown to be expressed on neuronal and glial cells of the human cerebrum, suggesting that EV-A71 is capable of invading the cerebrum through interactions with these receptors [85]. It has also been shown that EV-A71 can be isolated from brain parenchyma and spinal cord specimens, suggesting that the virus can be transported along peripheral nerves to the central nervous system via axonal nerve conduction [87]. In some cases, infants infected with EV-A71 may experience a range of neurological symptoms, such as aseptic meningitis, brain stem encephalitis, and other nervous system diseases. This suggests that EV-A71 may be capable of invading the central nervous system (CNS) through multiple pathways, such as crossing the blood-brain barrier [88], retrograde axon trafficking [89], and “Trojan horse” invasion [90] (Fig. 1).

Spread of enterovirus A71 in the body. The virus spreads from the oral-gut axis/lungs to peripheral tissues via blood/lymphatic circulation after infection. Viruses can cross the blood-brain barrier, hijack white blood cells, or reverse axon transport into the central nervous system
Crossing the blood-brain barrier
The blood-brain barrier (BBB) protects the CNS from harmful pathogens in the blood, but EV-A71 may cross the BBB via the transcellular pathway or the paracellular pathway [91]. Viruses in the blood can bind to vascular endothelial cells in the brain and be released across the BBB into the CNS through what is called the “transcellular pathway”. Sun et al. [86] suggested a model in which EV-A71 entered the mouse brain by inducing BBB leakage, and they showed that the destruction of endothelial cells of cerebral vessels by EV-A71 also helps the virus cross the BBB into the CNS. The permeability and tight connection of the BBB are closely related. In the paracellular pathway, circulating viruses in the blood can cross the BBB in a way that crosses the tight junction between neighboring endothelial cells without the involvement of receptors [91]. Wang and colleagues [91] demonstrated that the VP1 protein reduces the number of tight junction proteins between endothelial cells, thereby enhancing EV-A71’s ability to invade the brain via the paracellular pathway.
Retrograde axonal transport
Retrograde axonal transport (RAT) is considered the main route of neurotransmission [89]. In human autopsy tissues and animal models, EV-A71 can be transmitted into the CNS through axon trafficking. For example, Sun et al. [29] found that, after infecting human tonsil cells, EV-A17 can enter the CNS through facial nerve fibers that innervate the tonsils, via axon transport. The motor pathway is considered to be the main route of reverse axonal transport for EV-A71. The virus actively replicates in skeletal muscles, infects motor neurons at neuromuscular junctions (NMJs) and subsequently employs RAT to invade the spinal cord and reach the brainstem [89, 92, 93].
“Trojan horse” invasion
Metaphorically, Trojan horse invasion is a strategy by which host cells are induced to allow viruses to enter tissues and organs that are otherwise protected by their immune system. Some components of the host cell can be used as a Trojan horse to combine with components of the virus, facilitating the delivery of viral components to organelles of the host cell. Chang et al. [90] performed functional anatomical analysis on viral pathogenesis in an oral NOD/SCID mouse model and found that EV-A71 may initially invade leukocytes and macrophages in the intestinal tract of orally infected mice, using them as a “Trojan horse” to spread to multiple organs, including the muscles, heart, and lungs, via blood or lymphatic circulation. Huang et al. [94] showed that the host UDP-glucose glycoprotein glucosyltransferase 1 (UGGT1) facilitates EV-A71 invasion by binding to the 3D polymerase of EV-A71, after which the reintegrated UGGT1 is redeployed from the endoplasmic reticulum to the cytoplasm to facilitate viral replication.
EV-A71 infection triggers an immune response and immune evasion
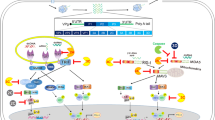
Viral infection can trigger various host immune responses to limit virus transmission, with the host’s innate antiviral immunity serving as the first line of defense against EV-A71 infection, capable of eradicating the invading virus. This innate immunity is also involved in the activation of adaptive immune responses, leading to a full range of immune protection. Cells in the human body contain innate receptors called pattern recognition receptors (PRRs) that can recognize pathogens as well as fragments of damaged or dead cells. When EV-A71 infection occurs, different PRRs detect EV-A71 viral RNA and trigger downstream signaling pathways that induce the production of type I Interferons and inflammatory cytokines. PRRs may be either intracellular or extracellular [33]. PRRs that have been shown to be capable of recognizing the EV-A71 virus include Toll-like receptors (TLRs) (TLR3, TLR7/8, and TLR9) and cytosolic RIG-I-like receptors (RLRs) (RIG-I and MDA5) [95]. TLRs are primarily responsible for detecting pathogen-associated molecular patterns (PAMPs) on the cell surface or within intracellular vesicles such as endosomes or lysosomes [96]. TLR3 recognizes viral dsRNA, while TLR7/8 recognizes viral ssRNA, and TLR9 recognizes DNA containing unmethylated 2’-deoxyribose (cytidine phosphoguanosine) (CpG) [97]. Unlike TLRs, cytoplasmic RLRs (RIG-1 and melanoma differentiation-associated gene 5 [MDA5]) detect viral RNA in the cytoplasm [98]. RIG-I recognizes short dsRNA and ssRNA with a 5’-triphosphate, while MDA5 recognizes long dsRNA [99]. Viruses have developed sophisticated strategies to subvert antiviral innate immunity by targeting the PRR pathway (including the TLR and RLR pathways) and the JAK-STAT pathway [100, 101] (Fig. 2). TLRs and RLRs have been shown to be involved in initiating a response against viral infection [33]. Interferons (IFNs) play an essential role in the antiviral immune response and are important for controlling the replication and spread of the virus [102]. IFNs are divided into three groups, based on their interaction with receptors: type I, type II, and type III. Type I and III interferons are thought to play an important role in defending against viral infections [103]. Type I IFNs (including IFNα and IFNβ) are the main cytokines that drive the antiviral defense in the early stages of viral infection and have two primary functions: First, IFNs generated by the TLR pathway and RLR pathway activate more than 300 IFN-stimulated genes (ISGs) via the JAK-STAT pathway, establishing an antiviral state in both virus-infected cells and neighboring cells (Fig. 2) [104, 105]. Type I IFNs induced by the PRR pathway bind to heterodimeric receptors consisting of interferon-α receptors 1 (IFNAR1) and 2 (IFNAR2), causing transphosphorylation and activation of tyrosine kinases such as TYK2 and JAK1. Phosphorylated STAT1/2 subsequently associates with IFN regulatory factor 9 (IRF9) to form a heterotrimeric complex, which is translocated to the nucleus and binds to IFN-stimulated response elements (ISREs) to activate the transcription of ISGs. Second, they promote dendritic cell maturation and enhance antigen presentation on T cells, which leads to a viral-antigen-specific adaptive immune response [106]. Together, the expression patterns of EV-A71 suggest that it can suppress the antiviral response early in infection that would inhibit its replication at the site of primary infection [107]. Type III IFNs play a more important role in local intestinal mucosal immunity. EV-A71 infection mainly induces the intestinal epithelium to produce IFN-λ, and IFN-λR (a heterodimer composed of IL-28Rα and IL-10Rβ chains) is highly expressed on the surface of intestinal mucosa, allowing infected cells of the intestinal epithelium to be recognized and killed by CD3+ intraepithelial lymphocytes (iIELs) and intestinal NK cells (iNK) [108].

Innate immunity and immune evasion by enterovirus A71. PRRs that recognize EV-A71 include TLRs (TLR3, TLR7/8, and TLR9) and cytosolic RLRs (RIG-I and MDA5), which bind to viral components to trigger innate antiviral immune responses in the TLR pathway and the RLR pathway, respectively, mainly producing type I IFNs. IFN, in turn, binds to its homologous receptor, and signals through the JAK/STAT pathway, and induces hundreds of ISGs, thereby increasing type I interferon levels and establishing a stable antiviral state between cells. At the same time, these innate immune responses are also involved in the production of pro-inflammatory factors. However, the virus achieves immune avoidance and downregulation of type I IFN by cleaving key proteins involved in the innate immune response through 2A protease, 2C protease, and 3C protease. MDA5, melanoma-differentiation-associated protein5; JAK1, Janus kinase 1; TYK2, tyrosine kinase 2; IFNAR, interferon-α/β receptor; MyD88, myeloid differential protein-88; TLR, Toll-like receptor; IRF9, interferon regulatory factor 9; IRF3/7, interferon regulatory factor 3/7; STAT1/2, signal transducer and activator of transcription 1/2; TRIF, TIR-domain-containing adapter-inducing interferon β; TAK1, transforming growth factor β activated kinase 1; TAB2/3, TGF-β activated kinase 2/3; IFNAR, interferon-α/β receptor; TBK1, TANK-binding kinase 1; RIG-I, retinoic-acid-inducible gene 1; MAVS, mitochondrial antiviral-signaling protein; IKK, inhibitor of kappa B kinase; NF-κB, nuclear factor kappa B; IkBα, NF kappa B inhibitor alpha; PP1, protein phosphatase-1; 2Apro, 2A protein; 2Cpro, 2C protein; 3Cpro, 3C protein
EV-A71 interferes with antiviral immune responses mainly through immune evasion. The 3C and 2A proteases of EV-A71 are the main antagonists of type I IFNs. EV-A71 3Cpro [109] and 2Apro [110] inhibit innate immunity by binding to and cleaving different antiviral signaling molecules, thereby affecting numerous cellular functions and shutting off cellular gene expression at the transcriptional and translational levels through interaction between key cell signaling molecules and viral proteins at various levels (Fig. 2). The viral proteins 2C [111] and 3D [112] play important roles in innate immune evasion. EV-A71 2C interacts with protein phosphatase 1 (PP1), recruits PP1 to IKKβ (inhibitor of kappa B kinase β) to form a 2C-PP1-IKKβ complex that inhibits IKKβ phosphorylation and, as a consequence, the nuclear factor-kappa B (NF-κB) signaling pathway [113]. 3D inhibits IFN-γ (gamma interferon) signaling, which may confer anti-EV-A71 activity [112]. The mechanism by which 3D contributes to immune escape is still unclear, and more research is needed to investigate this process. In addition, several microRNAs that are regulated by EV-A71 infection have been shown to be associated with immune evasion, for example, by evading host IL-6R (interleukin)- and STAT3 (signal transducer and activator of transcription3)-mediated antiviral activity [114, 115].
Apoptosis and inflammatory response pathways
There is increasing evidence that EV-A71 infection induces apoptosis in a range of cell lines, including HeLa, rhabdomyosarcoma (RD), Jurkat, SK-N-MC, glioblastoma SF268, Vero, and human microvascular endothelial cells [116,117,118,119,120,121]. The apoptotic pathway induced by EV-A71 appears to be cell-type-specific, resulting in caspase-dependent intrinsic apoptosis or calcium-induced caspase-independent apoptosis (Fig. 3). Proteases 2A, 3C, and 2B have been found to induce apoptosis, with the mechanism involving caspase activation. Notably, interactions between the EV-A71 2B protein and the innate immune system are thought to be important for mitochondrial apoptosis, as infection triggers conformational changes in the Bax protein, leading to apoptosis [30]. Sun et al. [29] demonstrated that EV-A71 infection in human tonsillar epithelial cells leads to apoptosis, which involves the release of cytochrome C from the mitochondria into the cytoplasm, activation of caspase 9, and a reduction in the number of apoptotic cells due to selective inhibition of caspase 9. These observations support the hypothesis that the mitochondria-mediated intrinsic apoptosis pathway in EV-A71-infected cells is a form of inflammatory programmed cell death. Ca2+ also plays a vital role in the initiation of apoptosis. The EV-A71 VP1 protein activates calmodulin-dependent protein kinase II (CaMKII) on the mitochondrial surface, which results in Ca2+ influx and elicits apoptosis-inducing factor (AIF), ultimately inducing apoptosis. Autophagy, especially in neuronal cells, may also lead to apoptosis associated with EV-A71 infection, and this is an important pathway of host defense against viruses [30].

EV-A71-induced inflammatory response pathway and apoptotic pathway. The EV-A71-triggered apoptosis pathway mainly includes the caspase-dependent mitochondrial apoptosis pathway and apoptosis caused by Ca2+ overload. Excessive inflammation can also lead to cell damage. The inflammatory response pathways mainly rely on EGFR to trigger MAPK signaling pathways, including the c-Src/EGFR/MEK/ERK and c-Src/PDGFR/PI3K/Akt/p42/p44 pathways. In addition, EV-A71 infection leads to the accumulation of intracellular and mitochondrial ROS, which can directly or indirectly activate JNK1/2, p38, ERK1/2, and NF-κB, further regulating the production of various pro-inflammatory cytokines. EGF, epidermal growth factor; c-Src, c-src tyrosine kinase; PDGFR, platelet-derived growth factor receptor; PIK3, phosphoinositide kinase 3; Akt, protein kinase B; P42/44:P42/44 mitogen-activated protein kinase; MAPK, mitogen-activated protein kinase; ERK, extracellular regulated kinase; MEX, MAPK/ERK kinase; JNK, c-Jun N-terminal kinase; COX-2, cyclooxygenase 2; PGE2, prostaglandin E2; p38, p38 MAP kinase; NLRP3, NLR family pyrin domain-containing 3; AIM2, absent in melanoma 2; Casp-1, caspase 1; Casp-9, caspase 9; AIF, apoptosis-inducing factor; Cyto c, cytochrome C; Bax, BCL2-associated X protein; CaMKII, Calcium-CaM-dependent protein kinase II
The inflammatory response produces pro-apoptotic cytokines, and therefore, the mechanism of apoptosis can partly be attributed to the inflammatory response to EV-A71 [35]. Clinical results suggest that deregulated inflammatory responses, such as cytokine storms, may play a key role in the pathogenesis of EV-A71 [33]. Studies have shown [2, 122, 123] that the following cytokines and chemokines are induced by EV-A71 infection: IL-1β, IL-2, IL-4, IL-8, IL-10, IL-12, IL-13, IL-17F, IL-18, IL-22, IL-23, IL-27, IL-33, IL-35, IL-37, TNF-α, IFN-γ, IP-10, MCP-1, G-CSF, and HMGB1. Many cellular pathways have been reported to be involved in EV-A71-induced inflammatory responses [35,36,37,38,39,40,41,42], with the mitogen-activated protein kinase (MAPK) signaling pathway playing a major role. Researchers have identified six MAPK subfamilies in mammalian cells, including p38MAPK (p38α /β/γ/δ), ERK1/2, ERK3/4, ERK7/8, JNK1/2/3, and ERK5/big MAP kinase 1 (BMK1) [124]. Epidermal growth factor receptor (EGFR) is present in cell membranes [125], and EGFR-dependent signaling is critical in inflammation pathogenesis due to its ability to regulate pro-inflammatory genes [34]. For example, EV-A71 infection triggers the overexpression of cyclooxygenase 2 (COX-2) and prostaglandin E2 (PGE2) via the c-Src/EGFR/MEK/ERK and c-Src/PDGFR/PI3K/Akt/p42/p44MAPK signaling pathways (Fig. 3). It is known that oxidative stress (OS) occurs when the production of reactive oxygen species (ROS) overwhelms cellular antioxidant defenses, and EV-A71-infection-induced ROS production can activate multiple inflammatory signaling pathways. During EV-A71 infection, integrin β1/EGFR signaling and mitochondrial damage lead to the accumulation of ROS, which then directly or indirectly activates JNK1/2, p38, ERK1/2, and nuclear factor kappa B (NF-κB) and further regulate the production of various pro-inflammatory cytokines (Fig. 3).
Moreover, EV-A71 stimulates the formation of intracellular inflammasome complexes, which promotes the production of pro-inflammatory factors. Recent studies [33, 126] have shown that AIM2, a cytoplasmic DNA sensor used to detect DNA viruses and other pathogens, is highly expressed in the central nervous system tissues of human EV-A71 encephalomyelitis patients. AIM2 can form an inflammasome by recruiting ASCs and procaspase 1 to trigger IL-1 maturation and pyroptosis (Fig. 3), but the underlying mechanisms by which EV-A71 infection activates AIM2 and the role of AIM in vivo during EV-A71 infection remain unclear. In addition, the cyclic inflammasome complex formed by NLRP3, EV-A71 3D, and ASC can also have a comparable function [127] (Fig. 3).
Therapeutic strategies and mechanisms
Currently, there are no effective antiviral drugs for the prevention, control, and treatment of severe EV-A71 infection. The drugs available for clinical treatment include nonspecific antiviral drugs such as IFN and ribavirin. Although new drugs, such as vemurafenib, flavonoids, and nucleic acid aptamer, offer advantages, their application is restricted due to a lack of in vivo experimental and clinical data (Table 1). In addition, potential drugs for the treatment of EV-A71 infection currently also include virus inhibitors targeting viral capsid proteins, the RNA-dependent RNA polymerase (RdRp), the 2C protein, the IRES, the 3C protease, the 2A protease, and other key sites (Table 1). However, most of these drugs are still in the early stages of development, and it is necessary to accelerate the progress of in vivo studies to allow their clinical efficacy to be evaluated. At present, the best way to prevent HFMD is through the administration of an effective and safe vaccine.
Drugs
Targeting host cells
Interferon
With a broad-spectrum antiviral effect [128, 129], IFN is widely used in clinical practice, and IFN-α has been found to be effective against EV-A71 infection [130]. As mentioned above, the RNA of EV-A71 is recognized by and binds to PPRs, leading to the activation of IFN production. IFN activates the JAK-STAT pathway, upregulates the expression of interferon-stimulated genes (ISGs), and then induces the antiviral effects of downstream pathways [104, 105]. Since EV-A71 infection can easily enter the CNS through SCARB2 in the early stages to cause pathological damage, early IFN treatment can prevent severe nervous system inflammation [45, 46]. Therefore, the older the child and the more mature the immune system, the more effective IFN treatment becomes [131].
However, the efficacy of IFN in treating severe infections is limited due to its short half-life, which necessitates repeated injections. This may increase respiratory and heart rates, resulting in many adverse reactions, with flu-like symptoms being the most common [47, 131]. Furthermore, the complex IFN-induced signaling pathway makes it challenging to fully elucidate the downstream response caused by key ISG genes. Therefore, further studies are needed to understand the mechanism of IFN-mediated inhibition of EV-A71 virus replication.
Vemurafenib
Vemurafenib is a V-Raf murine sarcoma viral oncogene homolog B1 (BRAF) gene inhibitor that targets the MAPK pathway transduction against EV-A71 infection, offering the advantage of being fast and efficient. Hu et al. [52] proposed that vemurafenib competitively activates the MAPK pathway with EV-A71, thus inhibiting the downstream pathway activated by EV-A71 through MAPK, which leads to a reduction in the expression of COX-2 and the release of pro-inflammatory factors in the NF-κB pathway. This approach is expected to be used to prevent nervous system inflammatory damage caused by EV-A71. In addition, vemurafenib can promote the expression of endogenous growth genes and inhibit the JNK/p38 MAPK pathway. It can also promote mitogenesis and proliferation of host cells, which in turn disrupts the replication of the viral genome. Vemurafenib is also known to inhibit pathways that promote apoptosis and inflammation, thus preventing severe disease associated with EV-A71 infection.
In conclusion, vemurafenib shows promising antiviral activity against EV-A71 infection, with greater efficacy observed when it is administered early. However, the complexity of the MAPK pathway and the exact mechanism by which vemurafenib induces viral genome damage and VP0 cleavage are still poorly understood, making it difficult to predict the potential interactions and links between in vivo factors and proteins. Therefore, current research on this topic remains limited to in vitro testing.
Targeting EV-A71
Ribavirin
Ribavirin, which has been approved by the US Food and Drug Administration (FDA), is a nonspecific antiviral drug that mainly works by inserting itself into RNA molecules of nascent viruses as a mutagenic agent. It binds to RNA polymerase, competitively inhibiting the synthesis of viral RNA [132]. In addition, ribavirin can inhibit de novo guanine synthesis by inhibiting the activity of hypoxanthine dehydrogenase (IMPDH). Thus, it reduces the concentration of guanosine triphosphate (GTP), thereby inhibiting viral RNA replication and protein translation and reducing the infectivity of the virus specifically [48, 132]. In terms of cellular effects, ribavirin can convert Th-2-type cells to Th-1-type cells, thereby affecting immune regulation and limiting the cell and tissue damage caused by antibodies and cytotoxic T lymphocytes (CTLs) [49, 50]. In 2019, Tate et al. [132] reported that ribavirin can also limit the replication, translation, packaging, and protease functions of DNA and RNA viruses by inducing spermidine-spermine N1-acetyltransferase 1 (SAT1) to break down polyamines, to which enteroviruses show a degree of sensitivity. Therefore, further studies on the relationship between mutagens and polyamine pathways are expected to provide new targets for designing broad-spectrum antiviral drugs. This suggests that combining ribavirin with drugs that induce the polyamine pathway can enhance the effectiveness of antiviral therapy.
However, ribavirin has the potential to inhibit the proliferation and differentiation of erythroid cells and megakaryocytes and can result in anorexia, diarrhea, immunosuppression, and other adverse reactions [133]. It may also increase mitochondrial toxicity, which leads to oxidative stress damage. Another potential concern is ribavirin’s antagonistic D1 receptor activity, which may cause adverse reactions affecting a patient’s mental state and neurobehavioral disorders [134,135,136]. Therefore, in the clinical use of ribavirin, special attention should be given to the specific condition of the patient, and further studies are needed to develop ways to prevent these adverse reactions.
Capsid inhibitors
Enterovirus capsid inhibitors were already being developed as early as the last century, and their development has progressed very quickly. However, their narrow antiviral spectrum remains a drawback. Although many drugs have been developed as viral protein capsid inhibitors, none of them have been put into clinical use (Table 1). Viral capsid inhibitors stabilize the capsid and prevent the release of the viral genome by replacing pocket factors with compounds with higher binding affinity for the conserved hydrophobic pocket (the canyon region on the capsid). One of these, lactoferrin, has the advantage of high efficiency and non-toxicity, while pyridine-based imidazolidinone compounds have become the most effective candidate drugs, targeting the virus through continuous modification and optimization, addressing the issue of resistance to certain strains.
Nucleic acid aptamers are short DNA or RNA molecules that are capable of recognizing specific receptors on cell membranes in vitro after undergoing selection and amplification via systematic evolution of ligands by exponential enrichment (SELEX), and this technique is expected to be utilized for targeted drug delivery due to its high affinity, targeting specificity, and cellular internalization capability [137, 138]. Aptamers not only acquire biological information about the target but also bind to the target molecule in a similar way to antibodies, anchoring on its surface without causing changes within the molecule [139]. They can therefore improve immunotherapy by enhancing specificity for the target molecule. VP1, an important structural protein of EV-A71, has been identified as a target for aptamers [140]. In 2021, Zou et al. [53] used SELEX to screen three DNA aptamers that could attach to the VP1 of EV-A71, and in a later in vitro antiviral test, they observed a significant reduction in the degree of cytopathic effect (CPE) in EV-A71-infected RD cells treated with aptamers, together with a notable decrease in the expression level of VP1, indicating the potential of aptamers to inhibit EV-A71 infection by targeting VP1. Although the exact mechanism by which they function remains to be investigated, aptamers appear to be promising candidates for combination therapy against EV-A71 infection due to their specificity for EV-A71-related targets [53] and their ability to bind to a variety of molecules, including nanoparticles, siRNA/miRNA, and chemical drugs [137, 138]. However, the current application of SELEX is limited to in vitro settings, and the nucleic acid sequence selection procedure is complicated. Long oligonucleotides tend to self-hybridize, while short oligonucleotides have lower specificity, and the mutability of EV-A71 makes it difficult to select optimized nucleic acid sequences [53]. Overcoming these problems is crucial for the future development of aptamer-based drugs to treatment of EV-A71 infections.
Protease and protein inhibitors
Increasing knowledge about the structural characteristics of EV-A71 proteins has led to the development of diverse inhibitors specific for different viral targets, including the IRES and the 3D, 2C, 3C, and 2A proteins (Table 1). Although their overall in vitro inhibitory is not as strong as that of capsid inhibitors, few of these compounds have been eliminated as a result of resistance selection tests. Currently, there are relatively few drugs targeting structural proteins such as VP4 and non-structural proteins such as 3A, and further development would be worthwhile.
3C proteases play an important role in promoting viral replication, inhibiting host innate immunity and triggering programmed cell death. For example, 3C proteases not only participate in the processing of most protein precursors but also affect Toll-like receptor (TLR), RIG-like receptor, Nod-like receptor (NLR) family PYRIN domains containing omega-3 (NLRP3), IFN, and other related signaling pathways by hydrolyzing host proteins [141]. Rupintrivir (AG7088), a peptide mimic inhibitor of 3C proteases containing a lactam group at P1, a fluorophenylalanine at P2, a Val at P3, a 5-methyl-3-isooxazole at P4, and an α, β-unsaturated ester at P1, is able to interact with EV-A71 3C and inhibit the function of 3C proteases through an extensive network of hydrogen bonds and hydrophobic interactions [142]. Rupintrivir (AG7088) is of particular research interest because of its ability to target 3C proteases. However, despite its promise for the treatment of severe diseases, rupintrivir has not yet been put to use, due to a lack of clinical safety trials and poor oral availability [143,144,145].
Because of the high sequence conservation of the 2C protein and its ability to reshape the structure of viral RNA and regulate the interaction between proteins through its helicase activity, inhibition of this activity can inhibit viral replication. Inhibitors of the 2C protein are considered promising candidates due to their broad-spectrum antiviral activity [145], and the drugs under investigation include designable peptides and quinolone-based dibucaine derivatives, which target the helicase activity. 2C ligand (2CL), which does not affect the type I response or cytokine pathways but instead competes with the C-terminal amphipathic helix of 2C for access to “the deep pocket” (a structure formed by the ATPase and zinc finger domains of another 2C protein), effectively inhibits the 2C helicase activity of a variety of viruses, including EV-A71, and disrupts the oligomerization of the 2C protein [146]. 2CL was also found to have a protective effect against fatal damage in newborn mice infected with EV-A71. Derivatives of dibucaine, such as 6aa (benzene modification) and 6af (thiophene modification) were originally designed as antiviral agents against EV-D68. Due to their high selectivity index and strong antiviral activity, a variety of optimized compounds such as 6i (N-ring modification) [145] and 6aw (fluorine-substituted benzene modification) [147] can also inhibit the helicase activity of the 2C protein. Therefore, the virus replication function mediated by the 2C protein is inhibited, and the antiviral effect can be exerted in the early stage. In addition, the introduction of fluorine improves the microsomal stability of 6aw, lengthening its half-life, which is conducive to the continuous action of the drug. Notably, derivatives of dibucaine, which also have oral activity, have shown strong antiviral activity with low cytotoxicity in studies in mice, leading to fewer side effects while protecting EV-A71-infected mice from fatal damage. Nevertheless, the activity of 2C protein inhibitors against EV-A71 is generally low. This is possibly related to the time of action of the drug at the target, so the stability of the drug in the liver microsome needs to be considered when designing 2C protein inhibitors against EV-A71.
Targeting both host cells and EV-A71
Flavonoids
Flavonoids, which are widely found in plants, share similar structures and functions such as anti-inflammatory, anti-cancer, antioxidant, and anti-allergic effects. Of these, apigenin has been shown to be the most effective in preventing cytotoxicity induced by EV-A71. In a study by Lv et al. [39], EV-A71 was found to be sensitive to apigenin, and a dose-dependent inhibition of peptide translation from the IRES of the 5’-UTR and blocking of EV-A71 genomic RNA synthesis were observed. In addition, apigenin was found to inhibit multiple responses induced by EV-A71 [39]. For example, it was found to inhibit the phosphorylation of components of the JNK/p38 MAPK pathway, leading to a reduction in viral replication. It was also found to block caspase-3 cleavage and to downregulate ROS to protect cells from ROS-induced damage. Apigenin also exerts anti-inflammatory effects by inhibiting the expression of most cytokines, except GM-CSF and IL-2. Although the specific mechanism of its inhibitory effect is not known, the number and position of hydroxyl groups in the B-ring of flavonoids may play an important role in the inhibition of IRES activity. Further research is necessary to examine the chemical structure and action mechanism of these flavonoids and their relationships [39].
in vivo experiments reported by Dai et al. [51] demonstrated for the first time that all flavonoids were effective in protecting neonatal mice from lethal EV-A71 challenges. This discovery highlights their potential as noncytotoxic antiviral drugs against EV-A71. However, the relative scarcity of in vivo data prevents a comprehensive understanding of the molecular mechanisms involved and the long-term toxicity of purified flavonoids, thus hindering their clinical approval for treating HFMD caused by EV-A71. It therefore is imperative that further safety studies be conducted.
Vaccine
As mentioned above, there is currently no specific treatment for HFMD, and the best way to prevent it is the use of effective and safe vaccines. Different types of vaccines have different mechanisms of action and specific advantages and disadvantages. It is important to note that a major problem with current EV-A71 vaccines is that they are generally subtype-specific and do not induce widespread neutralizing activity against all EV-A71 subtypes (Table 2).
Inactivated vaccine
Inviragen (Singapore) [57] and the National Health Research Institutes (NHRI, Taiwan) [58] have developed and put into clinical trials inactivated vaccines targeting HFMD infections caused by different subgenotypes of EV-A71. Encouragingly, three inactivated EV-A71 vaccines from Beijing Sinovac (China) [54], Beijing Vigoo (China) [55], and the Chinese Academy of Medical Science (CAMS, China), respectively [56], have been put on the market in China. Despite differences in vaccine strains, cell systems, development techniques, and virulence effects, the vaccines all achieve similar immunogenicity to the virus and can induce a specific immune response. Previous studies [148,149,150] have shown that inactivated vaccines can induce specific immune responses without causing strong immune-related inflammatory reactions through the regulation of various genes. Liang et al. [148] found that inoculation with the CAMS vaccine resulted in a strong Th1/Th2 response. The Th1 response delayed the inflammatory response, while the Th2 response induced the production of long-lasting specific neutralizing antibodies. This is often considered an important indicator of the post-immunization immune response.
In clinical studies, the inactivated EV-A71 vaccine has been found to be effective and safe [54,55,56, 151,152,153]. It is capable of producing specific long-lasting neutralizing antibodies [54, 56, 151,152,153], which can specifically prevent HFMD caused by EV-A71 infection and related serious complications [55, 151, 153], and it has the potential for cross-neutralization [152], which can help prevent hospitalization and severe cases. However, it also has some shortcomings that should not be ignored. First, the inactivated vaccine can cause local injection reactions such as redness, swelling, and pain at the injection site (sequelae have not yet been observed) because it requires the aluminum adjuvant to assist its function in vivo [54, 56, 151,152,153]. Second, young patients are more likely to suffer from adverse reactions, such as fever and local injection reactions [56, 152, 153], and it is worth noting that EV-A71 infections are most common in children under the age of 5. In addition, it is still not clear whether children who are given inactivated EV-A71 vaccine will produce cross-neutralization reactions when they are routinely vaccinated.
Live attenuated vaccine
Compared to inactivated vaccines, live attenuated vaccines can induce stronger humoral and cellular immunity, which means a longer-lasting immune state. In 2019, Ye et al. [59] developed a miRNA-based attenuated live vaccine strain (pIY) that showed reduced viral replication in both RD and SHYSY-5Y cells by inserting two miRNA target genes, let-7a and miR-124a, into specific locations in the genome of an EV-A71 mutant. The virus expressing the corresponding miRNA is not able to replicate in cells carrying the specific miRNA because the corresponding homologous miRNAs are present in these two cell lines. Moreover, pIY selectively inhibits its entry into skeletal muscle and the spinal cord, thus preventing serious neurological complications and reducing the viral load while retaining sufficient immunogenicity to induce a strong IFN-γ response and effective cellular immunity. This is conducive to the establishment of long-term memory and lifelong protection. Hence, pIY is considered a candidate vaccine for treating severe EV-A71 infection.
However, live attenuated vaccines also have limitations. For example, miRNA-based vaccines may produce escape mutant strains that have lower immunogenicity and have the potential to revert to virulence. Besides, the general safety of miRNA vaccines in humans remains to be evaluated.
In 2019, in order to de-optimize VP1 fragments, Tsai et al. [60] used synthetic attenuated virus engineering (SAVE) to insert codons of varying lengths into the capsid protein gene of a C4 genotype virus. This resulted in codon usage bias and an increase in CpG and UpA (uridine adenosine phosphate) dinucleotides, which affected the translation efficiency of viral proteins compared to the C2 genotype virus. Codon de-optimization preserves immunogenicity and reduces the efficiency of replication and translation of the dominant virus. This can reduce the virulence of EV-A71 and increase the neutralizing antibody titer. Furthermore, the high-fidelity determinant also ensures a low mutation rate and replication rate. This safe, stable, and up-to-date strategy has also been reflected in the development of vaccines against poliovirus and influenza A virus.
EV-A71-VLPs
An EV-A71 vaccine based on virus-like particles (EV-A71-VLPs) has become a new vaccine candidate with high levels of safety and effectiveness. Using a yeast system [154], Yang et al. [61] developed an EV-A71-VLPs vaccine against P1 produced in Pichia pastoris, which induced higher titers of neutralizing antibodies at high doses than the same amount of inactivated vaccine. In that study, the reason for the stronger immunogenicity was unclear, but it was probably related to the amino acid sequence of P1. In addition, the insertion of different promoters enabled the recombinant yeast to express more proteases and structural proteins, and the EV-A71-VLPs were highly purified, which was conducive to eliciting high titers of neutralizing antibodies in mice [62]. Although obtaining purified EV-A71-VLPs is time-consuming, the procedure is relatively easy to scale up for mass production. EV-A71-VLPs mimic the natural conformation of the virus and retains enough specific epitopes to achieve high immunogenicity [154,155,156]. After injection, EV-A71-VLPs can trigger high-level production of specific IgG antibodies and a variety of cytokines, such as IFN-γ, IL-2, IL-4, and IL-6, and can therefore induce a stronger specific immune response than that induced by an equal amount of inactivated vaccine. However, the persistence of neutralizing antibodies induced by EV-A71-VLPs and the specificity of the Th1/Th2 immune response after vaccination are still unknown and need to be studied further [62].
Synthetic peptide vaccine (VP1-based)
Subunit vaccines do not contain genetic material, which makes them safe and non-infectious. This type of vaccine includes recombinant vaccines, such as peptides and proteins. VP1 is a major antigen that can induce neutralizing antibodies, and it is therefore considered an ideal antigen for subunit vaccines. In 2021, Lei et al. [63] produced a series of synthetic peptides corresponding to the VP1 protein, and immunization of EV-A71-infected mice with three of them resulted in a reduction in lesions in the muscle, intestine, and brain. According to previous studies, only a portion of the epitopes carried by macromolecular antigens can induce protective antibodies [157]. Therefore, designing synthetic peptide vaccines containing these epitopes can help to induce a highly targeted immune response while reducing vaccine side effects and adverse reactions.
Although peptide-based vaccines are relatively safe and highly specific, the immune response they induce is weak because of their low immunogenicity. If these difficulties can be overcome, the clinical potential of peptide vaccines will increase greatly, because of their advantages regarding side effects and safety.
Conclusion
EV-A71 virus infection has become a global public health concern because of regional and seasonal outbreaks that can place a major burden on public health. Although symptoms are generally mild, and the infection is self-limiting, if the initial infection is not controlled in time, extensive invasion of tissues and organs can occur, potentially causing damage due to inflammation. More importantly, EV-A71 is capable of causing serious neurological damage, especially in infants and young children. This damage can manifest as aseptic meningitis, polio syndrome, and even death if nerve centers are infected.
The pathogenic mechanism of EV-A71 infection is complex, and a number of receptors have been reported to be involved in cell entry, including hSCARB2 [65, 66], HS proteoglycans [67], PSGL-1 [68], sialylated glycan [69, 70], Anx2 [71], vimentin [72], nucleolin [73], fibronectin [74], and prohibitin [12]. The main role of most of these receptors is to strengthen the adhesion of EV-A71 to the cell surface, but hSCARB2 is more specific and can also promote conformational changes in the virion that allow the viral RNA to be released in the cytoplasm. Blocking the binding of EV-A71 to attachment receptors is an important therapeutic strategy. In recent years, monoclonal antibodies have been developed for the treatment of EV-A71 infection, but this research is still at an early stage, and these antibodies are not yet in clinical use. Many factors contribute to the ability of EV-A71 to enter cells. PHBs and PRPH expressed on the cell surface can promote entry of the virus into nerve cells and aid in its replication [78, 158]. These factors are potential drug targets to limit neurological complications. Another factor, HSP90β, which is also expressed on the surface of the cell, facilitates viral entry by interacting with EV-A71 particles and protecting viral proteins from proteasomal degradation. After binding to the viral receptor, the virus enters the host cell through endocytosis and releases its positive-stranded RNA genome into the cytoplasm. The viral genomic RNA serves as an mRNA template for the translation of precursor polyproteins, which undergo further processing by virus-encoded proteases to produce functional viral proteins in infected cells.
If EV-A71 is not controlled or cleared soon after establishing an infection in the respiratory or gastrointestinal tract, it can spread to deep lymphoid tissue and replicate rapidly. This can eventually result in viremia in various tissues and organs through blood circulation. Importantly, intestinal organoids can secrete exosomes containing infectious virus particles, and these are also potential targets for inhibiting the spread of the virus within the body. It is worth noting that the location of the initial colonization site of EV-A71 is controversial, with some researchers suggesting that it is in lymphoid tissue of the oropharyngeal cavity (tonsils) and small intestine (Payer’s patch) [81, 85] and others suggesting that EV-A71 initially infects type II alveolar cells in the lungs [86]. The mechanism by which EV-A71 enters the CNS is also of great importance because this can lead to serious complications. It is currently thought that the virus infects the CNS by destroying endothelial cells and inducing BBB leakage, by retrograde axon transport, and by hijacking leukocytes (Fig. 1).
EV-A71 also subverts antiviral innate immunity by targeting the PRR and the JAK-STAT pathways (Fig. 2). In the PRR pathway, type I IFN-mediated antiviral immunity is triggered by different PRRs that detect EV-A71 viral RNA in various cell types. PRRs that recognize EV-A71 include TLRs (TLR3, TLR7/8 and TLR9) and cytosolic RLRs (RIG-I and MDA5) [95]. Type I IFNs, which are mainly produced by the PRR pathway, in turn trigger the intracellular JAK-STAT pathway, releasing more IFNs to act extracellularly on neighboring cells. As a result, a state of defense against the virus is established. However, the viral 3Cpro, 2Apro and 2Cpro have evolved the ability to exert immune avoidance by blocking key proteins in the innate immune response signaling pathway. There is also evidence suggesting that 3Dpro and several microRNAs also play important roles in innate immune evasion, but the mechanism by which this occurs is still unclear.
It is important to study the mechanisms involved in apoptosis and the inflammatory responses caused by EV-A71, as these damage tissues and organs (Fig. 3). EV-A71-induced apoptosis pathways include mitochondria-mediated intrinsic apoptosis pathways and non-caspase-dependent pathways caused by Ca2+ influx. Apoptosis is cell-specific. For example, the apoptotic mitochondrial pathway mediated by the activation and cleavage of caspase-9 has been identified as the main pathway through which EV-A71 induces the death of apoptotic cells and tonsillar epithelial cells [29, 117]. It is certain that the inflammatory response is antiviral. For example, a recent study [159] showed that IL-18 is protective against EV-A71 infection in mice and that the pathogenesis induced by EV-A71 infection can be reversed in vivo using recombinant IL-18. In contrast, an excessive inflammatory response can cause irreversible damage. The inflammatory response signaling pathway is mainly believed to be mediated primarily by the MAPK signaling pathway, and the accumulation of ROS in cells or mitochondria aggravates the inflammatory response (Fig. 3). In addition, the TLR and RLR signaling pathways produce other pro-inflammatory factors in addition to IFNs (Fig. 2).
Specific antiviral drugs for EV-A71 infection have not yet been developed. Nonspecific antiviral drugs such as IFN and ribavirin can be used for clinical treatment. The use of IFN against EV-A71 infection can activate ISG expression in the JAK-STAT pathway, inducing the innate immune response in the downstream pathway, blocking viral replication, inhibiting viral cytopathy, and preventing nervous system inflammation [45, 46, 104, 105, 160]. Ribavirin not only acts as a mutagen that increases the error rate of the viral polymerase and interferes with viral replication but also plays a role in immune regulation [48,49,50, 132]. The efficacy of IFN treatment is higher in older children than in younger children because their immune systems are more mature [131], and ribavirin is therefore more suitable for the treatment of young children with mild HFMD [161, 162]. Vemurafenib and flavonoids can also inhibit the JNK/p38 MAPK pathway and exert an anti-inflammatory effect, despite using different pathways to inhibit viral replication [39, 52, 163]. Vemurafenib acts quickly and effectively, but flavonoids are non-cytotoxic to neonates and are more beneficial for treating young children infected with EV-A71 [51, 52]. In addition, aptamer can enhance the specificity of target molecules associated with EV-A71 infection, making it a promising candidate for combination therapy [137,138,139]. Direct-acting EV-A71 antivirals such as 3C protease inhibitors and 2C protein inhibitors target a specific site to target 3Cpro and non-structural protein 2C, thus inhibiting replication and protein processing. Some peptides not only inhibit the function of various EV-A71 proteases but also inhibit the combination of EV-A71 and SCARB2. Due to their multiple targets, they may become promising drugs in the future. The current candidate drugs targeting the structure of EV-A71 are still in the early experimental stage or are still lacking in vivo or clinical trials, and there is still a long way to go before clinical application.
Vaccines can induce a specific immune response [59,60,61, 63]. An inactivated vaccine can specifically prevent EV-A71-associated HFMD and its severe complications by inducing a strong immune response without inducing a strong immune-related inflammatory response [55, 148,149,150,151, 153]. Live attenuated vaccines and EV-A71-VLPs can induce stronger specific immune responses than inactivated vaccines [59, 60, 154,155,156], and live attenuated vaccines can be used to avoid serious neurological complications [39, 40]. A synthetic peptide vaccine (such as a VP1-based vaccine) not only reduces the severity of lesions in the muscle, intestine, brain and other tissues but also minimizes the side effects and adverse reactions caused by unrelated immune responses [157]. The vaccines mentioned have different subtype specificities. The C4 strain may be the best candidate, as it has shown high cross-neutralization potential against all major EV-A71 genotypes and subgenotypes in clinical trials [55, 57, 58]. Considering that HFMD viruses include many genotypes and subgenotypes, our ultimate goal should be to focus on a multivalent HFMD vaccine against the main viruses involved.
Abbreviations
- HFMD:
-
hand, foot, and mouth disease
- EV-A71:
-
enterovirus A71
- RNA:
-
ribonucleic acid
- CV:
-
coxsackievirus
- PAMPs:
-
pathogen-associated molecular patterns
- SCARB2:
-
scavenger receptor B2
- PRRs:
-
pattern recognition receptors
- RD:
-
rhabdomyosarcoma
- UTR:
-
untranslated region
- eIF4E:
-
eukaryotic initiation factor 4E
- eIF3:
-
eukaryotic initiation factor 3
- RdRp:
-
RNA-dependent RNA polymerase
- PABP:
-
poly(A)-binding protein
- DNA:
-
deoxyribonucleic acid
- VP:
-
viral protein
- P1:
-
precursor protein 1
- hSCARB2:
-
human scavenger receptor class B, member 2
- HS:
-
heparan sulfate
- PSGL-1:
-
P-selectin glycoprotein ligand 1
- Anx2:
-
annexin II
- PHBs:
-
prohibitins
- HSP90β:
-
heat shock protein 90β
- GA:
-
geldanamycin
- 17-AAG:
-
analogue 17-allyamino-17-demethoxygeldanamycin
- HSP90:
-
heat shock protein 90
- CNS:
-
central nervous system
- PRPH:
-
peripherin
- BBB:
-
blood-brain barrier
- IFNAR:
-
interferon-α receptor
- RAT:
-
retrograde axonal transport
- NMJs:
-
neuromuscular junctions
- UGGT1:
-
UDP-glucose glycoprotein glucosyltransferase 1
- CpG:
-
cytidine phosphoguanosine
- MDA5:
-
melanoma differentiation associated gene 5
- IFN:
-
interferon
- ISG:
-
IFN-stimulated gene
- CaMKII:
-
calmodulin-dependent protein kinase II
- AIF:
-
apoptosis-inducing factor
- MAPK:
-
mitogen-activated protein kinase
- BMK1:
-
big MAP kinase 1
- EGFR:
-
epidermal growth factor receptor
- COX-2:
-
cyclooxygenase-2
- PGE2:
-
prostaglandin E2
- OS:
-
oxidative stress
- ROS:
-
reactive oxygen species
- NF-κB:
-
nuclear factor kappa B
- OAS3:
-
oligoadenylate synthetase 3
- IRF9:
-
IFN regulatory factor 9
- ISREs:
-
IFN-stimulated response elements
- iIELs:
-
intraepithelial lymphocytes
- iNK:
-
intestinal NK cells
- PP1:
-
protein phosphatase 1
- IKKβ:
-
inhibitor of kappa B kinase β
- FDA:
-
Food and Drug Administration
- IMPDH:
-
hypoxanthine dehydrogenase
- GTP:
-
guanosine triphosphate
- AIM:
-
absent in melanoma
- ASC:
-
apoptosis-associated speck-like protein containing CARD domain
- SAT1:
-
spermidine-spermine N1 acetyltransferase
- NLRP3:
-
NOD-like receptor thermal protein domain associated protein 3
- NLR:
-
NOD-like receptor
- 2CL:
-
2C ligand
- SELEX:
-
systematic evolution of ligands by exponential enrichment
- CPE:
-
cytopathic effect
- pIY:
-
miRNA-based attenuated live vaccine strain
- SAVE:
-
synthetic attenuated virus engineering
- VLPs:
-
virus-like particles
- IRES:
-
internal ribosome entry site
- BRAF:
-
V-Raf murine sarcoma viral oncogene homolog B1
- CTL:
-
cytotoxic T lymphocyte
- UpA:
-
uridine adenosine phosphate
References
Cox B, Levent F (2018) Hand, Foot, and Mouth Disease. JAMA 320:2492. https://doi.org/10.1001/jama.2018.17288
Gonzalez G, Carr MJ, Kobayashi M et al (2019) Enterovirus-Associated Hand-Foot and Mouth Disease and Neurological Complications in Japan and the Rest of the World. IJMS 20:5201. https://doi.org/10.3390/ijms20205201
Bruu A-L (2002) Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses and Newer Enteroviruses. In: Haaheim LR, Pattison JR, Whitley RJ (eds) A Practical Guide to Clinical Virology. John Wiley & Sons, Ltd, Chichester, UK, pp 44–45
Chavan NA, Lavania M, Shinde P et al (2023) The 2022 outbreak and the pathobiology of the coxsackie virus [hand foot and mouth disease] in India. Infect Genet Evol 111:105432. https://doi.org/10.1016/j.meegid.2023.105432
Messacar K, Abzug MJ, Dominguez SR (2016) The Emergence of Enterovirus-D68. https://doi.org/10.1128/microbiolspec.EI10-0018-2016. Microbiol Spectr 4:
Schmidt NJ, Lennette EH, Ho HH (1974) An Apparently New Enterovirus Isolated from Patients with Disease of the Central Nervous System. J Infect Dis 129:304–309. https://doi.org/10.1093/infdis/129.3.304
Chen K-T, Chang H-L, Wang S-T et al (2007) Epidemiologic Features of Hand-Foot-Mouth Disease and Herpangina Caused by Enterovirus 71 in Taiwan, 1998–2005. Pediatrics 120:e244–e252. https://doi.org/10.1542/peds.2006-3331
Cardosa MJ, Krishnan S, Tio PH et al (1999) Isolation of subgenus B adenovirus during a fatal outbreak of enterovirus 71-associated hand, foot, and mouth disease in Sibu, Sarawak. The Lancet 354:987–991. https://doi.org/10.1016/S0140-6736(98)11032-2
Chan KP, Goh KT, Chong CY et al (2003) Epidemic Hand, Foot and Mouth Disease Caused by Human Enterovirus 71, Singapore. Emerg Infect Dis 9
Chan LG, Parashar UD, Lye MS et al (2000) Deaths of Children during an Outbreak of Hand, Foot, and Mouth Disease in Sarawak, Malaysia: Clinical and Pathological Characteristics of the Disease. Clin Infect Dis 31:678–683. https://doi.org/10.1086/314032
Ho M, Chen E-R, Hsu K-H et al (1999) An Epidemic of Enterovirus 71 Infection in Taiwan. N Engl J Med 341:929–935. https://doi.org/10.1056/NEJM199909233411301
McMinn P, Stratov I, Nagarajan L, Davis S (2001) Neurological Manifestations of Enterovirus 71 Infection in Children during an Outbreak of Hand, Foot, and Mouth Disease in Western Australia. Clin Infect Dis 32:236–242. https://doi.org/10.1086/318454
Van Tu P, Thao NTT, Perera D et al (2007) Epidemiologic and Virologic Investigation of Hand, Foot, and Mouth Disease, Southern Vietnam, 2005. Emerg Infect Dis 13:1733–1741. https://doi.org/10.3201/eid1311.070632
Zhang Y, Tan X-J, Wang H-Y et al (2009) An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J Clin Virol 44:262–267. https://doi.org/10.1016/j.jcv.2009.02.002
Komatsu H, Shimizu Y, Takeuchi Y et al (1999) Outbreak of severe neurologic involvement associated with enterovirus 71 infection. Pediatr Neurol 20:17–23. https://doi.org/10.1016/S0887-8994(98)00087-3
Zhang Y, Zhu Z, Yang W et al (2010) An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of Hand Foot and Mouth Disease in Fuyang city of China. Virol J 7:1–9. https://doi.org/10.1186/1743-422X-7-94
M H (2000) Enterovirus 71: the virus, its infections and outbreaks. J Microbiol Immunol Infect 33:205–216
Gobara F, Itagaki A, Ito Y et al (1977) Properties of Virus Isolated from an Epidemic of Hand-Foot-and-Mouth Disease in 1973 in the City of Matsue: Comparison with Coxsackievirus Group A Type 16 Prototype. Microbiol Immunol 21:207–217. https://doi.org/10.1111/j.1348-0421.1977.tb00282.x
Hagiwara A, Tagaya I, Yoneyama T (1978) Epidemic of Hand, Foot and Mouth Disease Associated with Enterovirus 71 Infection. Intervirology 9:60–63. https://doi.org/10.1159/000148922
Gilbert GL, Dickson KE, Waters M-J et al (1988) Outbreak of enterovirus 71 infection in Victoria, Australia, with a high incidence of neurologic involvement: The Pediatric. Infect Disease J 7:484–487. https://doi.org/10.1097/00006454-198807000-00007
An L, Ga K, Va L, Mi M (2009) [Enterovirus 71: epidemiology and diagnostics]. Zhurnal mikrobiologii, epidemiologii i immunobiologii
Zhang Y, Tan X, Cui A et al (2013) Complete Genome Analysis of the C4 Subgenotype Strains of Enterovirus 71: Predominant Recombination C4 Viruses Persistently Circulating in China for 14 Years. PLoS ONE 8:e56341. https://doi.org/10.1371/journal.pone.0056341
Mizuta K, Aoki Y, Matoba Y et al (2014) Molecular epidemiology of enterovirus 71 strains isolated from children in Yamagata, Japan, between 1990 and 2013. J Med Microbiol 63:1356–1362. https://doi.org/10.1099/jmm.0.079699-0
Zhou J, Shi Y, Miao L et al (2021) Molecular epidemiology and recombination of Enterovirus A71 in mainland China from 1987 to 2017. Int Microbiol 24:291–299. https://doi.org/10.1007/s10123-021-00164-2
Huang Y-P, Lin T-L, Lin T-H, Wu H-S (2013) Antigenic and Genetic Diversity of Human Enterovirus 71 from 2009 to 2012, Taiwan. PLoS ONE 8:e80942. https://doi.org/10.1371/journal.pone.0080942
World Health Organization. Regional Office for the Western Pacific (2018) Hand, Foot and Mouth Disease Situation Update 2018. WHO Regional Office for the Western Pacific, Manila
Pallansch MA, Oberste MS, Whitton J (2013) Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. Fields Virol 490–530. https://doi.org/10.1002/0470857285.ch6
Takeuchi O, Akira S (2010) Pattern Recognition Receptors and Inflammation. Cell 140:805–820. https://doi.org/10.1016/j.cell.2010.01.022
Sun M, Yan K, Wang C et al (2021) Intrinsic apoptosis and cytokine induction regulated in human tonsillar epithelial cells infected with enterovirus A71. PLoS ONE 16:e0245529. https://doi.org/10.1371/journal.pone.0245529
Swain SK, Panda S, Sahu BP, Sarangi R (2022) Activation of Host Cellular Signaling and Mechanism of Enterovirus 71 Viral Proteins Associated with Hand, Foot and Mouth Disease. Viruses 14:2190. https://doi.org/10.3390/v14102190
Lu J-R, Lu W-W, Lai J-Z et al (2013) Calcium flux and calpain-mediated activation of the apoptosis-inducing factor contribute to enterovirus 71-induced apoptosis. J Gen Virol 94:1477–1485. https://doi.org/10.1099/vir.0.047753-0
Haolong C, Du N, Hongchao T et al (2013) Enterovirus 71 VP1 activates calmodulin-dependent protein kinase II and results in the rearrangement of vimentin in human astrocyte cells. PLoS ONE 8:e73900. https://doi.org/10.1371/journal.pone.0073900
Chen K-R, Ling P (2019) Interplays between Enterovirus A71 and the innate immune system. J Biomed Sci 26:95. https://doi.org/10.1186/s12929-019-0596-8
Wu W, Wages PA, Devlin RB et al (2015) Src-Mediated EGF Receptor Activation Regulates Ozone-Induced Interleukin 8 Expression in Human Bronchial Epithelial Cells. Environ Health Perspect 123:231–236. https://doi.org/10.1289/ehp.1307379
Lei X, Han N, Xiao X et al (2014) Enterovirus 71 3C Inhibits Cytokine Expression through Cleavage of the TAK1/TAB1/TAB2/TAB3 Complex. J Virol 88:9830–9841. https://doi.org/10.1128/JVI.01425-14
Leong SY, Ong BKT, Chu JJH (2015) The Role of Misshapen NCK-related kinase (MINK), a Novel Ste20 Family Kinase, in the IRES-Mediated Protein Translation of Human Enterovirus 71. PLoS Pathog 11:e1004686. https://doi.org/10.1371/journal.ppat.1004686
Xie G-C, Guo N-J, Grénman R et al (2016) Susceptibility of human tonsillar epithelial cells to enterovirus 71 with normal cytokine response. Virology 494:108–118. https://doi.org/10.1016/j.virol.2016.04.016
Madrid LV, Wang C-Y, Guttridge DC et al (2000) Akt Suppresses Apoptosis by Stimulating the Transactivation Potential of the RelA/p65 Subunit of NF-κB. Mol Cell Biol 20:1626–1638
Lv X, Qiu M, Chen D et al (2014) Apigenin inhibits enterovirus 71 replication through suppressing viral IRES activity and modulating cellular JNK pathway. Antiviral Res 109:30–41. https://doi.org/10.1016/j.antiviral.2014.06.004
Song J, Hu Y, Li J et al (2018) Suppression of the toll-like receptor 7-dependent type I interferon production pathway by autophagy resulting from enterovirus 71 and coxsackievirus A16 infections facilitates their replication. Arch Virol 163:135–144. https://doi.org/10.1007/s00705-017-3592-x
Du H, Yin P, Yang X et al (2015) Enterovirus 71 2C Protein Inhibits NF-κB Activation by Binding to RelA(p65). Sci Rep 5:14302. https://doi.org/10.1038/srep14302
Tung W-H, Lee I-T, Hsieh H-L, Yang C-M (2010) EV71 induces COX-2 expression via c-Src/PDGFR/PI3K/Akt/p42/p44 MAPK/AP-1 and NF-κB in rat brain astrocytes. J Cell Physiol 224:376–386. https://doi.org/10.1002/jcp.22133
Dang D, Zhang C, Zhang R et al (2017) Involvement of inducible nitric oxide synthase and mitochondrial dysfunction in the pathogenesis of enterovirus 71 infection. Oncotarget 8:81014–81026. https://doi.org/10.18632/oncotarget.21250
Zheng B, Zhou X, Tian L et al (2022) IFN-β1b induces OAS3 to inhibit EV71 via IFN-β1b/JAK/STAT1 pathway. Virol Sin 37:676–684. https://doi.org/10.1016/j.virs.2022.07.013
Liu M-L, Lee Y-P, Wang Y-F et al (2005) Type I interferons protect mice against enterovirus 71 infection. J Gen Virol 86:3263–3269. https://doi.org/10.1099/vir.0.81195-0
Huang CC, Liu CC, Chang YC et al (1999) Neurologic complications in children with enterovirus 71 infection. N Engl J Med 341:936–942. https://doi.org/10.1056/NEJM199909233411302
Sun J, Ennis J, Turner JD, Chu JJH (2016) Single dose of an adenovirus vectored mouse interferon-α protects mice from lethal EV71 challenge. Antiviral Res 134:207–215. https://doi.org/10.1016/j.antiviral.2016.09.003
Sadeghipour S, Bek EJ, McMinn PC (2013) Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J Virol 87:1759–1769. https://doi.org/10.1128/JVI.02139-12
Fang S-H, Hwang L-H, Chen D-S, Chiang B-L (2000) Ribavirin enhancement of hepatitis C virus core antigen-specific type 1 T helper cell response correlates with the increased IL-12 level. J Hepatol 33:791–798. https://doi.org/10.1016/S0168-8278(00)80312-8
Zuckerman E (2001) The effect of antiviral therapy on t(14;18) translocation and immunoglobulin gene rearrangement in patients with chronic hepatitis C virus infection. Blood 97:1555–1559. https://doi.org/10.1182/blood.V97.6.1555
Dai W, Bi J, Li F et al (2019) Antiviral Efficacy of Flavonoids against Enterovirus 71 Infection in Vitro and in Newborn Mice. https://doi.org/10.3390/v11070625. Viruses 11:
Hu B, Chik KK-H, Chan JF-W et al (2022) Vemurafenib Inhibits Enterovirus A71 Genome Replication and Virus Assembly. Pharmaceuticals (Basel) 15. https://doi.org/10.3390/ph15091067
Zou X, Wu J, Gu J et al (2021) DNA aptamer against EV-A71 VP1 protein: selection and application. Virol J 18:164. https://doi.org/10.1186/s12985-021-01631-y
Liu X, Yang W, Zhang C et al (2021) Immunogenicity and safety of an inactivated enterovirus 71 vaccine co-administered with measles-mumps-rubella vaccine and live-attenuated Japanese encephalitis vaccine: a phase 4, single-center, randomized controlled trial. Hum Vaccin Immunother 17:5348–5354. https://doi.org/10.1080/21645515.2021.2010428
Zhu F, Xu W, Xia J et al (2014) Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med 370:818–828. https://doi.org/10.1056/NEJMoa1304923
Guan X, Che Y, Wei S et al (2020) Effectiveness and Safety of an Inactivated Enterovirus 71 Vaccine in Children Aged 6–71 Months in a Phase IV Study. Clin Infect Dis 71:2421–2427. https://doi.org/10.1093/cid/ciz1114
Hwa S-H, Lee YA, Brewoo JN et al (2013) Preclinical evaluation of the immunogenicity and safety of an inactivated enterovirus 71 candidate vaccine. PLoS Negl Trop Dis 7:e2538. https://doi.org/10.1371/journal.pntd.0002538
Chang J-Y, Chang C-P, Tsai HH-P et al (2012) Selection and characterization of vaccine strain for Enterovirus 71 vaccine development. Vaccine 30:703–711. https://doi.org/10.1016/j.vaccine.2011.11.087
Yee PTI, Tan SH, Ong KC et al (2019) Development of live attenuated Enterovirus 71 vaccine strains that confer protection against lethal challenge in mice. Sci Rep 9:4805. https://doi.org/10.1038/s41598-019-41285-z
Tsai Y-H, Huang S-W, Hsieh W-S et al (2019) Enterovirus A71 Containing Codon-Deoptimized VP1 and High-Fidelity Polymerase as Next-Generation Vaccine Candidate. J Virol 93. https://doi.org/10.1128/JVI.02308-18
Yang Z, Gao F, Wang X et al (2020) Development and characterization of an enterovirus 71 (EV71) virus-like particles (VLPs) vaccine produced in Pichia pastoris. Hum Vaccin Immunother 16:1602–1610. https://doi.org/10.1080/21645515.2019.1649554
Kim H-J, Son HS, Lee SW et al (2019) Efficient expression of enterovirus 71 based on virus-like particles vaccine. PLoS ONE 14:e0210477. https://doi.org/10.1371/journal.pone.0210477
Lei L, Li Q, Xu S et al (2021) Transplantation of Enterovirus 71 Virion Protein Particle Vaccine Protects Against Enterovirus 71 Infection in a Neonatal Mouse Model. Ann Transpl 26:e924461. https://doi.org/10.12659/AOT.924461
Cox JA, Hiscox JA, Solomon T et al (2017) Immunopathogenesis and Virus–Host Interactions of Enterovirus 71 in Patients with Hand, Foot and Mouth Disease. Front Microbiol 8:2249. https://doi.org/10.3389/fmicb.2017.02249
Yamayoshi S, Yamashita Y, Li J et al (2009) Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med 15:798–801. https://doi.org/10.1038/nm.1992
Yamayoshi S, Ohka S, Fujii K, Koike S (2013) Functional Comparison of SCARB2 and PSGL1 as Receptors for Enterovirus 71. J Virol 87:3335–3347. https://doi.org/10.1128/JVI.02070-12
Tan CW, Poh CL, Sam I-C, Chan YF (2013) Enterovirus 71 Uses Cell Surface Heparan Sulfate Glycosaminoglycan as an Attachment Receptor. J Virol 87:611–620. https://doi.org/10.1128/JVI.02226-12
Nishimura Y, Shimojima M, Tano Y et al (2009) Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med 15:794–797. https://doi.org/10.1038/nm.1961
Su P-Y, Liu Y-T, Chang H-Y et al (2012) Cell surface sialylation affects binding of enterovirus 71 to rhabdomyosarcoma and neuroblastoma cells. BMC Microbiol 12:162. https://doi.org/10.1186/1471-2180-12-162
Yang B, Chuang H, Yang KD (2009) Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J 6:141. https://doi.org/10.1186/1743-422X-6-141
Yang B, Solakyildirim K, Chang Y, Linhardt RJ (2011) Hyphenated techniques for the analysis of heparin and heparan sulfate. Anal Bioanal Chem 399:541–557. https://doi.org/10.1007/s00216-010-4117-6
Du N, Cong H, Tian H et al (2014) Cell Surface Vimentin Is an Attachment Receptor for Enterovirus 71. J Virol 88:5816–5833. https://doi.org/10.1128/JVI.03826-13
Su P-Y, Wang Y-F, Huang S-W et al (2015) Cell Surface Nucleolin Facilitates Enterovirus 71 Binding and Infection. J Virol 89:4527–4538. https://doi.org/10.1128/JVI.03498-14
He Q, Ren S, Xia Z et al (2018) Fibronectin Facilitates Enterovirus 71 Infection by Mediating Viral Entry. J Virol 92:e02251–e02217. https://doi.org/10.1128/JVI.02251-17
Lin H-Y, Yang Y-T, Yu S-L et al (2013) Caveolar Endocytosis Is Required for Human PSGL-1-Mediated Enterovirus 71 Infection. J Virol 87:9064–9076. https://doi.org/10.1128/JVI.00573-13
Wang S, Pang Z, Fan H, Tong Y (2023) Advances in anti-EV-A71 drug development research. J Adv Res S2090-1232(23)00089–9. https://doi.org/10.1016/j.jare.2023.03.007
Bavelloni A, Piazzi M, Raffini M et al (2015) Prohibitin 2: At a communications crossroads. IUBMB Life 67:239–254. https://doi.org/10.1002/iub.1366
Lim ZQ, Ng QY, Oo Y et al (2021) Enterovirus-A71 exploits peripherin and Rac1 to invade the central nervous system. EMBO Rep 22. https://doi.org/10.15252/embr.202051777
Tsou Y-L, Lin Y-W, Chang H-W et al (2013) Heat Shock protein 90: Role in Enterovirus 71 Entry and Assembly and Potential Target for Therapy. PLoS ONE 8:e77133. https://doi.org/10.1371/journal.pone.0077133
Lee K-M, Wu C-C, Wu S-E et al (2020) The RNA-dependent RNA polymerase of enterovirus A71 associates with ribosomal proteins and positively regulates protein translation. RNA Biol 17:608–622. https://doi.org/10.1080/15476286.2020.1722448
Solomon T, Lewthwaite P, Perera D et al (2010) Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790. https://doi.org/10.1016/S1473-3099(10)70194-8
Song Y, Cheng X, Yang X et al (2015) Early growth response-1 facilitates enterovirus 71 replication by direct binding to the viral genome RNA. Int J Biochem Cell Biol 62:36–46. https://doi.org/10.1016/j.biocel.2015.02.012
Wang H, Li Y (2019) Recent Progress on Functional Genomics Research of Enterovirus 71. Virol Sin 34:9–21. https://doi.org/10.1007/s12250-018-0071-9
Huang H-I, Lin J-Y, Chiang H-C et al (2020) Exosomes Facilitate Transmission of Enterovirus A71 From Human Intestinal Epithelial Cells. J Infect Dis 222:456–469. https://doi.org/10.1093/infdis/jiaa174
Jiao X-Y, Guo L, Huang D-Y et al (2014) Distribution of EV71 receptors SCARB2 and PSGL-1 in human tissues. Virus Res 190:40–52. https://doi.org/10.1016/j.virusres.2014.05.007
Sun L, Tijsma A, Mirabelli C et al (2019) Intra-host emergence of an enterovirus A71 variant with enhanced PSGL1 usage and neurovirulence. Emerg Microbes Infections 8:1076–1085. https://doi.org/10.1080/22221751.2019.1644142
Liu K, Ma Y, Zhang C et al (2012) [Neurologic complications in children with enterovirus 71-infected hand-foot-mouth disease: clinical features, MRI findings and follow-up study]. Zhonghua Yi Xue Za Zhi 92:1742–1746
Zhao T, Zhang Z, Zhang Y et al (2017) Dynamic Interaction of Enterovirus 71 and Dendritic Cells in Infected Neonatal Rhesus Macaques. Front Cell Infect Microbiol 7:171. https://doi.org/10.3389/fcimb.2017.00171
Chen C-S, Yao Y-C, Lin S-C et al (2007) Retrograde Axonal Transport: a Major Transmission Route of Enterovirus 71 in Mice. J Virol 81:8996–9003. https://doi.org/10.1128/JVI.00236-07
Chang C-S, Liao C-C, Liou A-T et al (2019) Enterovirus 71 targets the cardiopulmonary system in a robust oral infection mouse model. Sci Rep 9:11108. https://doi.org/10.1038/s41598-019-47455-3
Wang W, Sun J, Wang N et al (2020) Enterovirus A71 capsid protein VP1 increases blood–brain barrier permeability and virus receptor vimentin on the brain endothelial cells. J Neurovirol 26:84–94. https://doi.org/10.1007/s13365-019-00800-8
Chen Y-C, Yu C-K, Wang Y-F et al (2004) A murine oral enterovirus 71 infection model with central nervous system involvement. J Gen Virol 85:69–77. https://doi.org/10.1099/vir.0.19423-0
Khong WX, Yan B, Yeo H et al (2012) A Non-Mouse-Adapted Enterovirus 71 (EV71) Strain Exhibits Neurotropism, Causing Neurological Manifestations in a Novel Mouse Model of EV71 Infection. J Virol 86:2121–2131. https://doi.org/10.1128/JVI.06103-11
Huang P-N, Jheng J-R, Arnold JJ et al (2017) UGGT1 enhances enterovirus 71 pathogenicity by promoting viral RNA synthesis and viral replication. PLoS Pathog 13:e1006375. https://doi.org/10.1371/journal.ppat.1006375
Pathinayake PS, Hsu AC-Y, Wark PAB (2015) Innate Immunity and Immune Evasion by Enterovirus 71. Viruses 7:6613–6630. https://doi.org/10.3390/v7122961
Lei X, Xiao X, Wang J (2016) Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction. Viruses 8:22. https://doi.org/10.3390/v8010022
Xagorari A, Chlichlia K (2008) Toll-Like Receptors and Viruses: Induction of Innate Antiviral Immune Responses. TOMICROJ 2:49–59. https://doi.org/10.2174/1874285800802010049
Kato H, Takeuchi O, Sato S et al (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. https://doi.org/10.1038/nature04734
Hornung V, Ellegast J, Kim S et al (2006) 5’-Triphosphate RNA Is the Ligand for RIG-I. Science 314:994–997. https://doi.org/10.1126/science.1132505
García-Sastre A (2017) Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 22:176–184. https://doi.org/10.1016/j.chom.2017.07.012
Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8:911–922. https://doi.org/10.1038/nri2436
Kontsek P (1994) Human type I interferons: structure and function. Acta Virol 38:345–360
Reid E, Charleston B (2014) Type I and III Interferon Production in Response to RNA Viruses. J Interferon Cytokine Res 34:649–658. https://doi.org/10.1089/jir.2014.0066
Kawai T, Akira S (2011) Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 34:637–650. https://doi.org/10.1016/j.immuni.2011.05.006
Chow J, Franz KM, Kagan JC (2015) PRRs are watching you: Localization of innate sensing and signaling regulators. Virology 479–480:104–109. https://doi.org/10.1016/j.virol.2015.02.051
McNab F, Mayer-Barber K, Sher A et al (2015) Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. https://doi.org/10.1038/nri3787
Kuo R-L, Chen Y-T, Li H-A et al (2021) Molecular determinants and heterogeneity underlying host response to EV-A71 infection at single-cell resolution. RNA Biol 18:796–808. https://doi.org/10.1080/15476286.2021.1872976
Dong Y, Liu J, Lu N, Zhang C (2021) Enterovirus 71 Antagonizes Antiviral Effects of Type III Interferon and Evades the Clearance of Intestinal Intraepithelial Lymphocytes. Front Microbiol 12:806084. https://doi.org/10.3389/fmicb.2021.806084
Weng K-F, Li M-L, Hung C-T, Shih S-R (2009) Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog 5:e1000593. https://doi.org/10.1371/journal.ppat.1000593
Wang T, Wang B, Huang H et al (2017) Enterovirus 71 protease 2Apro and 3Cpro differentially inhibit the cellular endoplasmic reticulum-associated degradation (ERAD) pathway via distinct mechanisms, and enterovirus 71 hijacks ERAD component p97 to promote its replication. PLoS Pathog 13:e1006674. https://doi.org/10.1371/journal.ppat.1006674
Wang S-H, Wang K, Zhao K et al (2020) The Structure, Function, and Mechanisms of Action of Enterovirus Non-structural Protein 2C. Front Microbiol 11:615965. https://doi.org/10.3389/fmicb.2020.615965
Wang L-C, Chen S-O, Chang S-P et al (2015) Enterovirus 71 Proteins 2A and 3D Antagonize the Antiviral Activity of Gamma Interferon via Signaling Attenuation. J Virol 89:7028–7037. https://doi.org/10.1128/JVI.00205-15
Zheng Z, Li H, Zhang Z et al (2011) Enterovirus 71 2C protein inhibits TNF-α-mediated activation of NF-κB by suppressing IκB kinase β phosphorylation. J Immunol 187:2202–2212. https://doi.org/10.4049/jimmunol.1100285
Chang Z, Wang Y, Bian L et al (2017) Enterovirus 71 antagonizes the antiviral activity of host STAT3 and IL-6R with partial dependence on virus-induced miR-124. J Gen Virol 98:3008–3025. https://doi.org/10.1099/jgv.0.000967
Feng N, Zhou Z, Li Y et al (2017) Enterovirus 71-induced has-miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. Virus Res 237:27–36. https://doi.org/10.1016/j.virusres.2017.05.008
Li M-L, Hsu T-A, Chen T-C et al (2002) The 3C Protease Activity of Enterovirus 71 Induces Human Neural Cell Apoptosis. Virology 293:386–395. https://doi.org/10.1006/viro.2001.1310
Chang S-C, Lin J-Y, Lo LY-C et al (2004) Diverse apoptotic pathways in enterovirus 71–infected cells. J Neurovirol 10:338–349. https://doi.org/10.1080/13550280490521032
Liang C-C, Sun M-J, Lei H-Y et al (2004) Human endothelial cell activation and apoptosis induced by enterovirus 71 infection. J Med Virol 74:597–603. https://doi.org/10.1002/jmv.20216
Chen L-C, Shyu H-W, Chen S-H et al (2006) Enterovirus 71 infection induces Fas ligand expression and apoptosis of Jurkat cells. J Med Virol 78:780–786. https://doi.org/10.1002/jmv.20623
Wang S, Lei H, Huang K et al (2003) Pathogenesis of Enterovirus 71 Brainstem Encephalitis in Pediatric Patients: Roles of Cytokines and Cellular Immune Activation in Patients with Pulmonary Edema. J INFECT DIS 188:564–570. https://doi.org/10.1086/376998
Chen T-C, Lai Y-K, Yu C-K, Juang J-L (2007) Enterovirus 71 triggering of neuronal apoptosis through activation of Abl-Cdk5 signalling. Cell Microbiol 9:2676–2688. https://doi.org/10.1111/j.1462-5822.2007.00988.x
Wang Y, Qin Y, Wang T et al (2018) Pyroptosis induced by enterovirus 71 and coxsackievirus B3 infection affects viral replication and host response. Sci Rep 8:2887. https://doi.org/10.1038/s41598-018-20958-1
Chang L-Y (2004) Transmission and Clinical Features of Enterovirus 71 Infections in Household Contacts in Taiwan. JAMA 291:222. https://doi.org/10.1001/jama.291.2.222
Krishna M, Narang H (2008) The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 65:3525–3544. https://doi.org/10.1007/s00018-008-8170-7
Polosa R, Sapsford RJ, Dokic D et al (2004) Induction of the epidermal growth factor receptor and its ligands in nasal epithelium by ozone. J Allergy Clin Immunol 113:120–126. https://doi.org/10.1016/j.jaci.2003.09.040
Yogarajah T, Ong KC, Perera D, Wong KT (2017) AIM2 Inflammasome-Mediated Pyroptosis in Enterovirus A71-Infected Neuronal Cells Restricts Viral Replication. Sci Rep 7:5845. https://doi.org/10.1038/s41598-017-05589-2
Wang W, Xiao F, Wan P et al (2017) EV71 3D Protein Binds with NLRP3 and Enhances the Assembly of Inflammasome Complex. PLoS Pathog 13:e1006123. https://doi.org/10.1371/journal.ppat.1006123
Itsui Y, Sakamoto N, Kurosaki M et al (2006) Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat 13:690–700. https://doi.org/10.1111/j.1365-2893.2006.00732.x
Long L, Wu L, Chen L et al (2021) Effect of early oxygen therapy and antiviral treatment on disease progression in patients with COVID-19: A retrospective study of medical charts in China. PLoS Negl Trop Dis 15:e0009051. https://doi.org/10.1371/journal.pntd.0009051
Li X-W, Ni X, Qian S-Y et al (2018) Chinese guidelines for the diagnosis and treatment of hand, foot and mouth disease (2018 edition). World J Pediatr 14:437–447. https://doi.org/10.1007/s12519-018-0189-8
Lin H, Huang L, Zhou J et al (2016) Efficacy and safety of interferon-α2b spray in the treatment of hand, foot, and mouth disease: a multicenter, randomized, double-blind trial. Arch Virol 161:3073–3080. https://doi.org/10.1007/s00705-016-3012-7
Tate PM, Mastrodomenico V, Mounce BC (2019) Ribavirin Induces Polyamine Depletion via Nucleotide Depletion to Limit Virus Replication. Cell Rep 28:2620–2633e4. https://doi.org/10.1016/j.celrep.2019.07.099
Noshy MM, Hussien NA, El-Ghor AA (2013) Evaluation of the role of the antioxidant silymarin in modulating the in vivo genotoxicity of the antiviral drug ribavirin in mice. Mutat Res 752:14–20. https://doi.org/10.1016/j.mrgentox.2012.12.012
Davoodi L, Masoum B, Moosazadeh M et al (2018) Psychiatric side effects of pegylated interferon–α and ribavirin therapy in Iranian patients with chronic hepatitis C: A meta–analysis. Exp Ther Med. https://doi.org/10.3892/etm.2018.6255
Dieperink E, Ho SB, Thuras P, Willenbring ML (2003) A Prospective Study of Neuropsychiatric Symptoms Associated With Interferon-α-2b and Ribavirin Therapy for Patients With Chronic Hepatitis C. Psychosomatics 44:104–112. https://doi.org/10.1176/appi.psy.44.2.104
Predescu O, Streba LA-M, Irimia E et al (2012) Adverse effects of peg-Interferon and Ribavirin combined antiviral treatment in a Romanian hepatitis C virus infected cohort. Rom J Morphol Embryol 53:497–502
Van Simaeys D, López-Colón D, Sefah K et al (2010) Study of the Molecular Recognition of Aptamers Selected through Ovarian Cancer Cell-SELEX. PLoS ONE 5:e13770. https://doi.org/10.1371/journal.pone.0013770
Tuerk C, Gold L (1990) Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 249:505–510. https://doi.org/10.1126/science.2200121
Yang S, Wen J, Li H et al (2019) Aptamer-Engineered Natural Killer Cells for Cell-Specific Adaptive Immunotherapy. Small 15:e1900903. https://doi.org/10.1002/smll.201900903
Li W, Feng X, Yan X et al (2016) A DNA Aptamer Against Influenza A Virus: An Effective Inhibitor to the Hemagglutinin-Glycan Interactions. Nucleic Acid Ther 26:166–172. https://doi.org/10.1089/nat.2015.0564
Wen W, Qi Z, Wang J (2020) The Function and Mechanism of Enterovirus 71 (EV71) 3C Protease. Curr Microbiol 77:1968–1975. https://doi.org/10.1007/s00284-020-02082-4
Lu G, Qi J, Chen Z et al (2011) Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J Virol 85:10319–10331. https://doi.org/10.1128/JVI.00787-11
Zhang X, Song Z, Qin B et al (2013) Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Res 97:264–269. https://doi.org/10.1016/j.antiviral.2012.12.029
Zhang X-N, Song Z-G, Jiang T et al (2010) Rupintrivir is a promising candidate for treating severe cases of Enterovirus-71 infection. World J Gastroenterol 16:201–209. https://doi.org/10.3748/wjg.v16.i2.201
Tang Q, Xu Z, Jin M et al (2020) Identification of dibucaine derivatives as novel potent enterovirus 2C helicase inhibitors: In vitro, in vivo, and combination therapy study. Eur J Med Chem 202:112310. https://doi.org/10.1016/j.ejmech.2020.112310
Fang Y, Wang C, Wang C et al (2021) Antiviral Peptides Targeting the Helicase Activity of Enterovirus Nonstructural Protein 2C. J Virol 95. https://doi.org/10.1128/JVI.02324-20
Musharrafieh R, Kitamura N, Hu Y, Wang J (2020) Development of broad-spectrum enterovirus antivirals based on quinoline scaffold. Bioorg Chem 101:103981. https://doi.org/10.1016/j.bioorg.2020.103981
Liang Y, Zhou X, Yang E et al (2012) Analysis of the Th1/Th2 Reaction in the Immune Response Induced by EV71 Inactivated Vaccine in Neonatal Rhesus Monkeys. J Clin Immunol 32:1048–1058. https://doi.org/10.1007/s10875-012-9690-3
Liu L, Zhang Y, Wang J et al (2013) Study of the integrated immune response induced by an inactivated EV71 vaccine. PLoS ONE 8:e54451. https://doi.org/10.1371/journal.pone.0054451
Wu C-Y, Lin Y-W, Kuo C-H et al (2015) Inactivated Enterovirus 71 Vaccine Produced by 200-L Scale Serum-Free Microcarrier Bioreactor System Provides Cross-Protective Efficacy in Human SCARB2 Transgenic Mouse. PLoS ONE 10:e0136420. https://doi.org/10.1371/journal.pone.0136420
Cheng A, Fung C-P, Liu C-C et al (2013) A Phase I, randomized, open-label study to evaluate the safety and immunogenicity of an enterovirus 71 vaccine. Vaccine 31:2471–2476. https://doi.org/10.1016/j.vaccine.2013.03.015
Tambyah PA, Oon J, Asli R et al (2019) An inactivated enterovirus 71 vaccine is safe and immunogenic in healthy adults: A phase I, double blind, randomized, placebo-controlled, study of two dosages. Vaccine 37:4344–4353. https://doi.org/10.1016/j.vaccine.2019.06.023
Zhang L, Gao F, Zeng G et al (2021) Immunogenicity and Safety of Inactivated Enterovirus 71 Vaccine in Children Aged 36–71 Months: A Double-Blind, Randomized, Controlled, Non-inferiority Phase III Trial. J Pediatr Infect Dis Soc 10:440–447. https://doi.org/10.1093/jpids/piaa129
Wang Z, Zhou C, Gao F et al (2021) Preclinical evaluation of recombinant HFMD vaccine based on enterovirus 71 (EV71) virus-like particles (VLP): Immunogenicity, efficacy and toxicology. Vaccine 39:4296–4305. https://doi.org/10.1016/j.vaccine.2021.06.031
Zhang C, Ku Z, Liu Q et al (2015) High-yield production of recombinant virus-like particles of enterovirus 71 in Pichia pastoris and their protective efficacy against oral viral challenge in mice. Vaccine 33:2335–2341. https://doi.org/10.1016/j.vaccine.2015.03.034
Li H-Y, Han J-F, Qin C-F, Chen R (2013) Virus-like particles for enterovirus 71 produced from Saccharomyces cerevisiae potently elicits protective immune responses in mice. Vaccine 31:3281–3287. https://doi.org/10.1016/j.vaccine.2013.05.019
Kim Y-G, Lee Y, Kim JH et al (2020) Self-Assembled Multi-Epitope Peptide Amphiphiles Enhance the Immune Response against Enterovirus 71. Nanomaterials (Basel) 10:. https://doi.org/10.3390/nano10122342
Andino R, Böddeker N, Silvera D, Gamarnik AV (1999) Intracellular determinants of picornavirus replication. Trends Microbiol 7:76–82. https://doi.org/10.1016/S0966-842X(98)01446-2
Li Z, Wang H, Chen Y et al (2017) Interleukin-18 protects mice from Enterovirus 71 infection. Cytokine 96:132–137. https://doi.org/10.1016/j.cyto.2017.04.002
Guo X, Zeng S, Ji X et al (2022) Type I Interferon-Induced TMEM106A Blocks Attachment of EV-A71 Virus by Interacting With the Membrane Protein SCARB2. Front Immunol 13:817835. https://doi.org/10.3389/fimmu.2022.817835
Zhang H-P, Wang L, Qian J-H et al (2014) [Efficacy and safety of ribavirin aerosol in children with hand-foot-mouth disease]. Zhongguo Dang Dai Er Ke Za Zhi 16:272–276
Pan S, Qian J, Gong X, Zhou Y (2014) [Effects of ribavirin aerosol on viral exclusion of patients with hand-foot-mouth disease]. Zhonghua Yi Xue Za Zhi 94:1563–1566
Zhang W, Qiao H, Lv Y et al (2014) Apigenin inhibits enterovirus-71 infection by disrupting viral RNA association with trans-acting factors. PLoS ONE 9:e110429. https://doi.org/10.1371/journal.pone.0110429
Acknowledgments
The authors thank Professor Lingqing Xu from the Department of Qingyuan People’s Hospital, who carefully reviewed the manuscript and provided his expert opinion.
Authors’ contributions
Funding
This paper was not funded.
Author information
Authors and Affiliations
Contributions
Jianmei Lai and Zhishan Li designed the study and drafted the manuscript. Lixin Pan and Chunhong Ma collected data. Yunxia Huang and Jiachun Guo analyzed the data. Jianmei Lai, Zhishan Li, and Zifei Zhou refined the manuscript and coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this article. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Communicated by Ana Cristina Bratanich
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jianmei Lai and Zhishan Li contributed equally to this work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lai, J., Li, Z., Pan, L. et al. Research progress on pathogenic and therapeutic mechanisms of Enterovirus A71. Arch Virol 168, 260 (2023). https://doi.org/10.1007/s00705-023-05882-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05882-8